






基因组变异注释 — ANNOVAR(一)
1 简介
ANNOVAR 是一款高效的基因组注释工具,专门用于分析和注释来自多种生物基因组(包括人类的 hg18、hg19、hg38,以及小鼠、蠕虫、果蝇、酵母等)的遗传变异。这个工具实际上就是几个用 Perl 语言编写的脚本,因此可以在安装了 Perl 解释器的多种操作系统上(一般 Linux 自带)运行。
ANNOVAR 的核心功能在于其多样的注释方法,包括基于基因的注释(gene-based annotation)、基于区域的注释(region-based annotation)和基于筛选的注释(filter-based annotation)。基于基因的注释可以识别单核苷酸多态性(SNPs)或拷贝数变异(CNVs)是否导致蛋白质编码的变化及其影响的氨基酸;基于区域的注释用于鉴定特定基因组区域内的变异,如保守区域、转录因子结合位点、GWAS 候选区间等;而基于筛选的注释则用于判断变异是否被特定数据库(如dbSNP、1000 Genome项目)记录,以及进行多种功能预测评分,这也是 ANNOVAR 的主要优势之一。

ANNOVAR 支持多种输入和输出格式,最常见的输入格式是 VCF 文件,而输出则包括注释过的 VCF 文件、以tab 或逗号分隔的文本文件。除此之外,ANNOVAR 还提供了其他功能,如批量获取特定基因组位置的核苷酸序列,为孟德尔病从外显子组数据中识别候选基因列表等。
总而言之,ANNOVAR 是一款功能丰富、灵活高效的基因组注释工具,适用于广泛的基因组研究和遗传变异分析。与之类似的变异注释软件还包括 VEP、snpEff、VAAST、AnnTools 等。
扫码关注微信公众号【生信F3】获取更多生物信息学最新知识。

2 安装
-
访问官网下载页面:ANNOVAR Download Center.
-
注册
在下载页面填写基本信息并使用机构邮箱(.edu 或 .gov 结尾的邮箱地址)进行注册。
-
下载
完成注册后,ANNOVAR 会直接在当前页面刷新后提供链接。

Linux 系统下载 ANNOVAR 可使用以下命令,但实测下载速度很慢。
wget http://www.openbioinformatics.org/annovar/download/*/annovar.latest.tar.gz推荐使用 Motrix 在 windows 本地下载后上传至服务器中。
-
解压和安装
在 Linux 系统(一般自带 Perl)中,使用 tar 命令解压后即可使用:
tar -xvfz annovar.latest.tar.gz解压后的文件夹中包含以下文件:
-
example:存放的是示例数据文件 -
humandb:部分注释数据库的文件,annovar 的软件中默认自带了 hg19 的 refGene 数据库; -
annotate_variation.pl:主程序,用来进行数据库的下载,以及不同形式的注释; -
coding_change.pl:用来推断蛋白质的序列是否发生变化; -
convert2annovar.pl:将其他多种文件格式(VCF 等)转化为 annovar 可识别的形式; -
retrieve_seq_from_fasta.pl:自行建立其它物种的转录本 -
table_annovar.pl:可以一次完成三种不同形式的注释 -
variants_reduction.pl:用来定制过滤注释流程
-
3 用法
3.1 准备输入文件
convert2annovar.pl 脚本可以将其他 “genotype calling” 格式转换为 ANNOVAR 格式。目前,该程序可以处理Samtools 基因型 calling pileup 格式,Illumina CASAVA 格式,SOLiD GFF 基因型调用格式,Complete Genomics 变异格式,SOAPsnp 格式,MAQ 格式和 VCF 格式。此外,该程序还可以从 dbSNP 标识符列表、转录标识符或基因组区域生成 ANNOVAR 输入文件。现今,VCF 是使用最广泛的文件格式,而大多数其他格式已不再使用。
convert2annovar.pl \
-format vcf4 \
-allsample \
-outfile SV \
input.vcf
-
--formatvcf4输入文件格式(默认:pileup)
-
--outfilefile输出文件名(default: STDOUT)
-
--allsample对于多样本 VCF 文件,该参数将处理文件中的所有样本,并为每个样本生成单独的输出文件。默认情况下,只处理 VCF 文件中的第一个样本。
转换后用于后续 annovar 注释的输入文件格式为 .avinput,示例如下:
cat example/ex1.avinput
# 1 948921 948921 T C comments: rs15842, a SNP in 5' UTR of ISG15
# 1 1404001 1404001 G T comments: rs149123833, a SNP in 3' UTR of ATAD3C
# 1 5935162 5935162 A T comments: rs1287637, a splice site variant in NPHP4
# 1 162736463 162736463 C T comments: rs1000050, a SNP in Illumina SNP arrays
# 1 84875173 84875173 C T comments: rs6576700 or SNP_A-1780419, a SNP in Affymetrix SNP arrays
# 1 13211293 13211294 TC - comments: rs59770105, a 2-bp deletion
# 1 11403596 11403596 - AT comments: rs35561142, a 2-bp insertion
# 1 105492231 105492231 A ATAAA comments: rs10552169, a block substitution
# 1 67705958 67705958 G A comments: rs11209026 (R381Q), a SNP in IL23R associated with Crohn's disease
# 2 234183368 234183368 A G comments: rs2241880 (T300A), a SNP in the ATG16L1 associated with Crohn's disease
# 16 50745926 50745926 C T comments: rs2066844 (R702W), a non-synonymous SNP in NOD2
# 16 50756540 50756540 G C comments: rs2066845 (G908R), a non-synonymous SNP in NOD2
# 16 50763778 50763778 - C comments: rs2066847 (c.3016_3017insC), a frameshift SNP in NOD2
# 13 20763686 20763686 G - comments: rs1801002 (del35G), a frameshift mutation in GJB2, associated with hearing loss
# 13 20797176 21105944 0 - comments: a 342kb deletion encompassing GJB6, associated with hearing loss
# 8 8887543 8887543 A T comments: a mutation that abolishes stop codon
# 8 8887539 8887539 A T comments: a mutation that results in premature stop codon
# 8 8887536 8887537 AG GATT comments: a mutation that creates a stop codon 2 amino acids downstream
# 8 8887540 8887540 G GGAA comments: a mutation that results in insertion of a new amino acid
# 5 1295288 1295288 G A comments: a variant upstream of transcriptional start site
# chr14 95602958 95602958 A C comments: a variant that affects splicing of UTR regions
文件以空格或者制表符分隔,最少需要 5 列,分别代表:
-
染色体 (Chromosome)
-
起始位置 (Start)
-
终止位置 (End)
-
参考等位基因 (Reference Allele)
-
替代等位基因 (Alternative Allele)
-
其他的列作为额外补充信息 (可选)。插入或者删除以
-表示,“0” 代表只指定position,而不指定实际的核苷酸。
3.2 数据库下载
ANNOVAR 的注释主要依赖于数据库,因此在进行分析之前,应将所需的数据库下载到 humandb 文件夹中,下载的命令如下:
annotate_variation.pl -buildver hg19 -downdb -webfrom annovar avsnp147 humandb/
-buildver:对应参考基因组的版本
-downdb –webfrom annovar:从 ANNOVAR 库中下载对应的数据库,可获取的数据库查询网址为:(http://annovar.openbioinformatics.org/en/latest/user-guide/download/)
avsnp147:下载的数据库的名称,各个数据库的详细介绍可以参考以下链接:https://annovar.openbioinformatics.org/en/latest/user-guide/filter/
humandb:下载到 humandb 文件夹中
3.3 基因注释
确定 SNPs、InDels、SVs 或 CNVs 等是否会引起蛋白质编码变化以及受影响的氨基酸。用户可以灵活使用 RefSeq 基因、UCSC 基因、ENSEMBL 基因、GENCODE 基因、AceView 基因等多种基因定义系统。
3.3.1 模式物种变异注释
对于人类等模式生物,现在下载 ANNOVAR 的最新版本默认自带了 hg19 的数据库,所以基于 hg19 进行的变异检测可以很方便的直接注释SNV/InDel/SV/CNV,如果是 hg38 等其它基因组版本,则需自行下载后再进行注释。
annotate_variation.pl \
-geneanno \
-buildver hg19 \
-dbtype refGene \
-outfile SV.S1.anno \
-exonsort \
SV.S1.avinput /path/to/annovar/humandb
-buildver 基因组版本
-dbtype 数据库类型,参考:https://annovar.openbioinformatics.org/en/latest/user-guide/download/
-neargene [int] 定义基因上游/下游的距离阈值
-exonsort 结果按外显子排序
3.3.2 非模式物种变异注释
非模式物种
针对非模式物种需要首先自己构建数据库,准备基因组文件(genome.fasta)、基因组注释文件(gtf)
gtfToGenePred -genePredExt genome.gtf genome_refGene.txt
./annovar/retrieve_seq_from_fasta.pl \
--format refGene \
--seqfile genome.fasta \
--out genome_refGeneMrna.fa \
genome_refGene.txt
3.4 区间注释
识别特定基因组区域的变异,例如 44 个物种的保守区域,预测转录因子结合位点,片段重复区域,GWAS hits,基因组变异数据库,DNAse I超敏位点,ENCODE H3K4Me1/H3K4Me3/H3K27Ac/CTCF 位点,ChIP-Seq 峰,RNA-Seq 峰,或许多其他基因组区间的注释。
3.4.1 识别细胞遗传带(cytogenetic band)
要识别吉氏(Giemsa)染色带,需要首先下载对应数据库信息:
annotate_variation.pl -build hg19 -downdb cytoBand annovar/humandb/
可使用 -dbtype cytoBand 选项:
annotate_variation.pl -regionanno -buildver hg19 -dbtype cytoBand ex1.avinput humandb/
当一个变异跨越多个条带时,它们之间会用破折号连接(例如 1q21.1-q23.3)。
3.5 数据库注释(基于筛选的注释)
这个使用频率非常高,而且通常是结合多个数据库信息一起过滤。
重点是检查那些后缀为 dropped 的文件,Known variants will be written to the dropped file together with allele frequencies. 如下:
238104 tmp.hg38_ALL.sites.2015_08_dropped
15575 tmp.hg38_clinvar_20170905_dropped
7527 tmp.hg38_cosmic70_dropped
172432 tmp.hg38_exac03_dropped
281449 tmp.hg38_gnomad_genome_dropped
可以看到,总共的48万位点,其中有13万是在千人基因组计划出现的,有17万是在EXAC数据库出现,但是只有区区7527个位点是在COSMIC数据库出现,,在clinvar数据库的,有15575位点。
最原始的想法,通常是找到某个基因被注释到外显子区域的nonsynonymous突变位点,然后如果其被clinvar数据库记录,那就说明找到了有证据支持的致病位点。
不过,值得一提的是 clinvar 数据库需要经常更新哦。
~/biosoft/ANNOVAR/annovar/annotate_variation.pl -downdb clinvar_20180603 humandb -buildver hg38
# 72M Jul 9 2018 hg38_clinvar_20180603.txt
~/biosoft/ANNOVAR/annovar/annotate_variation.pl -filter \
-buildver hg38 -dbtype clinvar_20180603 --outfile tmp for_annovar.input \
~/biosoft/ANNOVAR/annovar/humandb/
3.6 联合注释
对于初学者来说,使用 ANNOVAR 的最简单方法是使用 table_annovar.pl 程序。该程序接收一个输入变异文件(如 VCF 文件),并生成一个以制表符分隔的输出文件,文件中有许多列,每列代表一组注释。此外,如果输入是 VCF 文件,程序还会生成一个新的输出 VCF 文件,并在 INFO 字段中填入注释信息。
使用 table_annovar.pl 主程序对 example/ex1.avinput 输入文件同时进行多数据库的注释:
# 下载refGene数据库
annotate_variation.pl -buildver hg19 -downdb -webfrom annovar refGene humandb/
# 下载refGene数据库
annotate_variation.pl -buildver hg19 -downdb cytoBand humandb/
# 下载refGene数据库
annotate_variation.pl -buildver hg19 -downdb -webfrom annovar exac03 humandb/
# 下载refGene数据库
annotate_variation.pl -buildver hg19 -downdb -webfrom annovar avsnp147 humandb/
# 下载refGene数据库
annotate_variation.pl -buildver hg19 -downdb -webfrom annovar dbnsfp30a humandb/
# 利用以上下载的数据库注释
table_annovar.pl example/ex1.avinput humandb/ \
-buildver hg19 \
-out myanno \
-remove \
-protocol refGene,cytoBand,exac03,avsnp147,dbnsfp30a \
-operation gx,r,f,f,f \
-nastring . \
-csvout \
-polish \
-xref example/gene_xref.txt
-
-protocol注释来源数据库的准确名称,有多少个数据库,需要与随后的 operation 方案一一对应。
-
-operation注释的类型:
g表示基于基因的注释(gene-based annotation)、r表示基于区域的注释(region-based annotation) 、f表示基于筛选的注释( filter-based annotation)。 -
-polish优化索引的蛋白质符号(如p.G12Vfs*2)
依次运行以上命令。前几个命令将适当的数据库下载到 humandb/ 目录中。最后运行 TABLE_ANNOVAR 命令,使用 ExAC 版本0.3 (简称exac03) dbNFSP版本3.0a (简称dbnsfp30a), dbSNP 版本147 使用左规范化(简称avsnp147) 数据库并删除所有临时文件,并生成名为 myanno.hg19_multianno.txt 的输出文件。没有任何注释的字段将由 . 字符串填充。在 Excel 中打开输出文件,查看其中包含的内容。
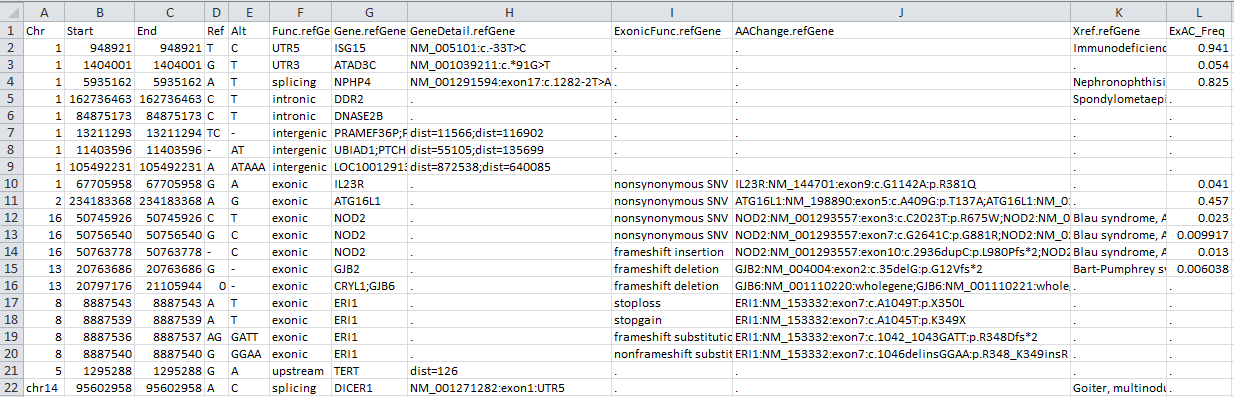
table_annovar.pl 可以直接支持 VCF 文件的输入和输出 (注释将写入输出 VCF 文件的 INFO 字段):
table_annovar.pl example/ex2.vcf humandb/ \
-buildver hg19 \
-out myanno \
-protocol refGene,cytoBand \
-operation g,r \
-nastring . -vcfinput -polish -remove
输出内容以两种方式展示:
-
ex2.hg19_multianno.vcf -
ex2. hg19_multiano .txt
其中包含不同格式的类似信息。
在得到变异位点的注释信息后,我们就可以按需要筛选我们感兴趣(如与疾病关联)的遗传位点了。关于如何筛选的内容请参考微信公众号【生信F3】中《致病基因分析详解》。
扫码关注微信公众号【生信F3】获取更多生物信息学最新知识。
本文由 mdnice 多平台发布

















 2645
2645

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









