






之前用Kraken2注释宏基因组的contig,发现只有30%左右可以被Kraken2注释
Kraken2+Bracken:宏基因组物种注释-CSDN博客
不信邪,再用NR库试试
参考:
将NR数据库diamond比对结果做物种注释_diamond 物种注释-CSDN博客
NR下载
nohup wget -t 0 -c -b https://ftp.ncbi.nlm.nih.gov/blast/db/FASTA/nr.gz &
wget -c https://ftp.ncbi.nlm.nih.gov/blast/db/FASTA/nr.gz.md5
#检查下载的完整性(一样的话就是完整了)
md5sum nr.gz ### 899ac219cac213c60fede9c3d9ef8f7b nr.gz
cat nr.gz.md5 ### 899ac219cac213c60fede9c3d9ef8f7b nr.gz
nohup gunzip nr.gz &
mv nr nr.fa
###下载NR物种相关信息和taxid信息
wget -t 0 -c -b https://ftp.ncbi.nlm.nih.gov/pub/taxonomy/accession2taxid/prot.accession2taxid.FULL.gz
wget -t 0 -c -b https://ftp.ncbi.nlm.nih.gov/pub/taxonomy/accession2taxid/prot.accession2taxid.FULL.gz.md5
wget -t 0 -c -b https://ftp.ncbi.nlm.nih.gov/pub/taxonomy/taxdump.tar.gz
wget -t 0 -c -b https://ftp.ncbi.nlm.nih.gov/pub/taxonomy/taxdump.tar.gz.md5
#检查完整性
md5sum prot.accession2taxid.FULL.gz
cat prot.accession2taxid.FULL.gz.md5
md5sum taxdump.tar.gz
cat taxdump.tar.gz.md5
#解压
tar -zxvf taxdump.tar.gz
gunzip prot.accession2taxid.gz
#移动需要的文件
cd ~
mkdir .taxonkit
cp *.dmp ~/.taxonkit
必要软件下载
conda create -n NR_database_search
conda activate NR_database_search
conda install -c bioconda taxonkit=0.15.0
conda install -c bioconda diamond=2.1.8
conda install -c bioconda csvtk=0.28.0diamond建库
nohup diamond makedb --threads 180 --in nr.fa --db NR_2023_07_23 &diamond比对
diamond blastx --db NR_2023_07_23.dmnd --query nucleic_reads.fna\
-o nucleic_matches_fmt6.txt --threads 180 --evalue 0.00001 \
--max-target-seqs 5 --outfmt 6
diamond blastp --db NR_2023_07_23.dmnd --query protein_reads.fna\
-o protein_matches_fmt6.txt --threads 180 --evalue 0.00001 \
--max-target-seqs 10000 --outfmt 6
## --outfmt 6 最好别改变这些参数可以调整diamond比对的速度or准确性
这几个参数可以调整比对的coverage,identity,score

结果如下 !!!(这个表头后面python会加)
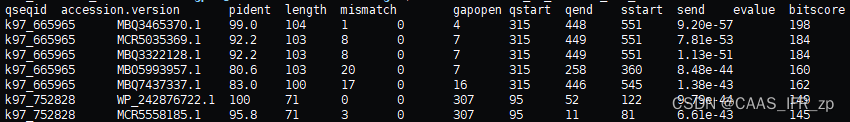
taxonkit获得物种分类信息表
感谢大佬:一文完成nt库序列快速下载及blast结果注释物种 (qq.com)
得到seqid注释之后,可以搜索注释
## 一些主要的物种编号(不含藻类和高级生物)
# 2 bacteria
# 2157 archaea
# 4751 fungi
# 10239 virus
# 554915 Amoebozoa
# 2686027 Ancyromonadida
# 554296 Apusozoa
# 1401294 Breviatea
# 2608240 CRuMs
# 2611352 Discoba
# 2489521 Hemimastigophora
# 2611341 Metamonada
# 2795258 Malawimonadida
# 3004206 Provora
# 2698737 SAR
#看taxnokit安好了么
taxonkit -h
#创建目录
cd /home/zhongpei/database/NR_2023_07_23
mkdir /home/zhongpei/database/NR_2023_07_23/NCBI_Main_tax
cd NCBI_Main_tax
#开始(构建不同的子库)
taxonkit list -j 4 --ids 2 --indent "" > NCBI_Main.taxid.bacteria.txt
taxonkit list -j 4 --ids 2157 --indent "" > NCBI_Main.taxid.archaea.txt
taxonkit list -j 4 --ids 4751 --indent "" > NCBI_Main.taxid.fungi.txt
taxonkit list -j 4 --ids 10239 --indent "" > NCBI_Main.taxid.virus.txt
taxonkit list -j 4 --ids 554915,2686027,554296,1401294,2608240,2611352,2489521,2611341,2795258,3004206,2698737 --indent "" > NCBI_Main.taxid.Eukaryotic.txt
# -j 是线程,软件说4个够了;--ids 是需要的物种编号,用逗号分隔
# 提取taxid和taxonomy(界门纲目科属种)的对应信息到NCBI_Main.taxid.txt
less NCBI_Main.taxid.bacteria.txt | taxonkit reformat -I 1 -r Unassigned -f "{k}\t{p}\t{c}\t{o}\t{f}\t{g}\t{s}\t{t}"| sed '1i\Taxid\tKingdom\tPhylum\tClass\tOrder\tFamily\tGenus\tSpecies\tStrain' > NCBI_Main.taxid.bacteria_new.txt
less NCBI_Main.taxid.archaea.txt | taxonkit reformat -I 1 -r Unassigned -f "{k}\t{p}\t{c}\t{o}\t{f}\t{g}\t{s}\t{t}"| sed '1i\Taxid\tKingdom\tPhylum\tClass\tOrder\tFamily\tGenus\tSpecies\tStrain' > NCBI_Main.taxid.archaea_new.txt
less NCBI_Main.taxid.fungi.txt | taxonkit reformat -I 1 -r Unassigned -f "{k}\t{p}\t{c}\t{o}\t{f}\t{g}\t{s}\t{t}"| sed '1i\Taxid\tKingdom\tPhylum\tClass\tOrder\tFamily\tGenus\tSpecies\tStrain' > NCBI_Main.taxid.fungi_new.txt
less NCBI_Main.taxid.virus.txt | taxonkit reformat -I 1 -r Unassigned -f "{k}\t{p}\t{c}\t{o}\t{f}\t{g}\t{s}\t{t}"| sed '1i\Taxid\tKingdom\tPhylum\tClass\tOrder\tFamily\tGenus\tSpecies\tStrain' > NCBI_Main.taxid.virus_new.txt
less NCBI_Main.taxid.Eukaryotic.txt | taxonkit reformat -I 1 -r Unassigned -f "{k}\t{p}\t{c}\t{o}\t{f}\t{g}\t{s}\t{t}"| sed '1i\Taxid\tKingdom\tPhylum\tClass\tOrder\tFamily\tGenus\tSpecies\tStrain' > NCBI_Main.taxid.Eukaryotic_new.txt
# -I 1 一个制表符分隔;-r 没有找到的用什么字符去填充,这里用的“Unassigned”
# -f 输出的格式;1i表示在第行之前插入文本(sed用法,不太会)
完成!
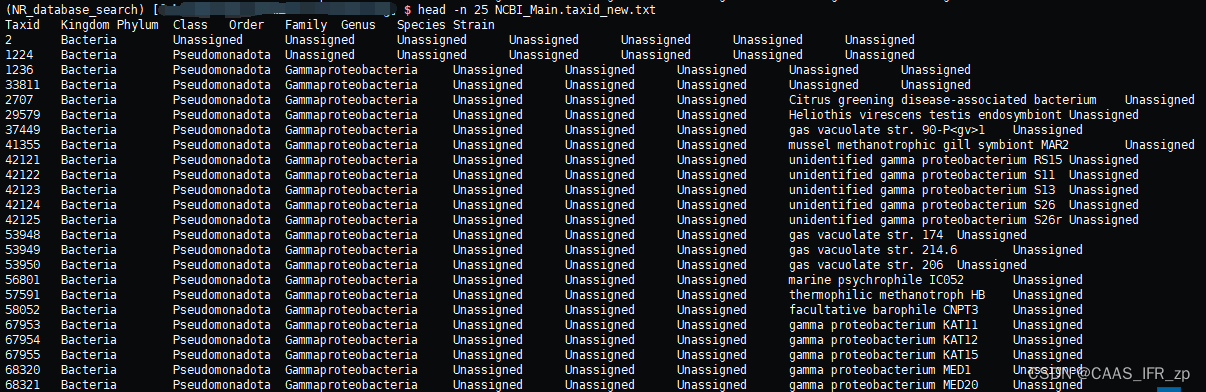
vim NCBI_Main.taxid_new.txt
把第一个Taxid改成小写
seqid和taxid的对应
还记不记得第一步下载过一个 "prot.accession2taxid" ,现在要派上用场了
其实python也能做,csvtk太不熟悉了,先来学习一下吧,感觉还挺方便(这一步比较慢)
cat prot.accession2taxid.FULL | csvtk -t grep -f taxid -P NCBI_Main.taxid.txt | csvtk -t cut -f accession.version,taxid > NCBI_seqid_taxid.txt
# "cat prot.accession2taxid |" 把 prot.accession2taxid 的内容到下面的 csvtk
# -t 输入内容是制表符分隔;grep 这是csvtk的1个子命令,用于在文件中搜索匹配的行
# -P 搜索那些"taxid"字段的值出现在"NCBI_Main.taxid.txt"文件中的行
# cut 这是csvtk的1个子命令,用于从输入中选择特定的字段
# 如果细菌的库太大了,可以分割
nohup split -l 2000000000 NCBI_seqid_taxid_bacteria.txt NCBI_seqid_taxid_bacteria_
NCBI_seqid_taxid.txt 就是目标文件
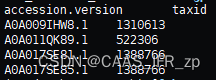
diamond seqid和taxid对应,再和界门纲目科属种对应
筛选diamond结果里面得分最高的(第一个出现的)
#!/usr/bin/env python
import argparse
parser = argparse.ArgumentParser(description="Filter input file to retain only the first occurrence of each qseqid.")
parser.add_argument("--input", "-i", required=True, help="Path to the input file.")
parser.add_argument("--output", "-o", required=True, help="Path to the output file.")
args = parser.parse_args()
input_file = args.input
output_file = args.output
seen_qseqids = set()
with open(input_file, 'r') as infile, open(output_file, 'w') as outfile:
first_line = infile.readline().strip()
if "qseqid" in first_line.lower():
outfile.write(first_line + '\n')
else:
parts = first_line.split('\t')
qseqid = parts[0]
if qseqid not in seen_qseqids:
outfile.write(first_line + '\n')
seen_qseqids.add(qseqid)
for line in infile:
parts = line.strip().split('\t')
qseqid = parts[0]
if qseqid not in seen_qseqids:
outfile.write(line)
seen_qseqids.add(qseqid)
print(f"Filtered file saved as '{output_file}'.")把diamond结果文件与NCBI_seqid_taxid.txt对应
#!/usr/bin/env python
# ##########################################################
# match diamond blast NR result.txt and NCBI_seqid_taxid.txt
# written by PeiZhong in IFR of CAAS
import argparse
import os
parser = argparse.ArgumentParser(description='Match Diamond BLAST NR result.txt and NCBI_seqid_taxid.txt. '
'!!! All files should end with .txt !!!')
parser.add_argument("--file_maker", "-m", nargs='+', required=True,
help="Keywords to filter input files (e.g., diamond txt)")
parser.add_argument("--input_dir", "-i", required=True,
help="Input directory containing result files")
parser.add_argument("--database", "-db", required=True,
help="File = NCBI_seqid_taxid.txt")
parser.add_argument("--out_name", "-o", required=True,
help="Output name for processed result files")
args = parser.parse_args()
file_makers = args.file_maker
input_dir = args.input_dir
database = args.database
out_name = args.out_name
db = {}
# Step 1: Read and build the database
print("Start reading database...")
with open(database, 'r') as f_table:
for line in f_table:
line = line.split('\t')
acc_num = line[0].strip()
tax_num = line[1].strip()
db[acc_num] = tax_num
print("Finished building database.")
# Step 2: Find files in input directory matching all makers
print(f"Scanning input directory: {input_dir}")
all_files = os.listdir(input_dir)
filtered_files = [
os.path.join(input_dir, file)
for file in all_files
if all(maker in file for maker in file_makers)
]
if not filtered_files:
print(f"No files matched the makers: {' '.join(file_makers)} in directory {input_dir}")
exit(1)
print(f"Found {len(filtered_files)} file(s) matching the criteria.")
# Step 3: Process each filtered file
for result in filtered_files:
print(f"Processing file: {result}")
# Add header to output file
header = "qseqid\taccession.version\ttaxid"
with open(result, 'r') as f_result, open(out_name, 'w') as f_out:
f_out.write(header + '\n')
# Match each line in the result file with the database
for line in f_result:
if "qseqid" in line: # Skip header
continue
line = line.strip().split('\t')
query_num = line[0].strip()
acc_q_num = line[1].strip()
# Check if the accession number exists in the database
if acc_q_num in db:
tax_id = db[acc_q_num]
else:
continue
f_out.write(f"{query_num}\t{acc_q_num}\t{tax_id}\n")
print(f"Finished processing file: {result}. Output saved to {out_name}")
print("All files processed.")chmod +x diamond_NR_tax.py(完整地址)
diamond_NR_tax.py(完整地址) -h
nohup /home/zhongpei/hard_disk_sda2/zhongpei/Software/my_script/diamond_NR_tax.py --file_maker diamond --input_dir /home/zhongpei/diarrhoea/fan/metagenomic/bin/CAT_bins --database NCBI_seqid_taxid.txt --outdir .
# 可以写个shell代码批量比对所有seqid_taxid文件
for i in NCBI_seqid_taxid_*.txt
do
num=${i%%.txt}
/home/zhongpei/hard_disk_sda2/zhongpei/Software/my_script/diamond_NR_tax.py -m diamond txt -i /home/zhongpei/diarrhoea/fan/metagenomic/assembly_result -db ${num}.txt -o ${num}_result.txt
done做完结果是这样的
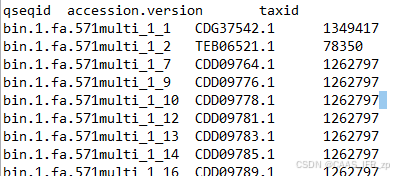
接下来再写个python代码根据taxid把这个文件和界门纲目科属种联系起来就行(不好意思只会python,我检讨。。。。但是python简单呀)
#!/usr/bin/env python
# ##########################################################
# Match Diamond BLAST NR taxid results and NCBI_Main.taxid_new.txt
# Written by PeiZhong in IFR of CAAS
import os
import argparse
# Step 1: Set up argument parser
parser = argparse.ArgumentParser(description='Match Diamond BLAST NR taxid result.txt and NCBI_Main.taxid_new.txt. '
'!!! All files should end with .txt !!!')
parser.add_argument('--input_txt', '-i', required=True,
help='path of the folder containing your seqid_taxid files')
parser.add_argument('--ncbi_db', '-db', required=True,
help='path to NCBI_Main.taxid_new.txt file')
parser.add_argument('--out', '-o', required=True,
help='Output directory to save results')
args = parser.parse_args()
input_txt = args.input_txt
ncbi_db = args.ncbi_db
out = args.out
# Step 1: Build the taxid database
print("Reading NCBI database...")
db = {}
with open(ncbi_db, 'r') as f_db:
for line in f_db:
line = line.strip().split('\t')
taxid = line[0].strip()
tax_info = line[1:8] # Extract additional taxonomic information
db[taxid] = tax_info
print(f"Database loaded: {len(db)} entries.")
# Step 2: Process result file
print(f"Processing file: {input_txt}")
with open(input_txt, 'r') as f_result, open(out, 'w') as f_out:
# Add header to output file
header = "qseqid\taccession.version\ttaxid\tkingdom\tphylum\tclass\torder\tfamily\tgenus\tspecies"
f_out.write(header + '\n')
for line in f_result:
line = line.strip().split('\t')
if len(line) < 3: # Skip lines with insufficient columns
continue
query_num = line[0].strip()
acc_q_num = line[1].strip()
taxid_1 = line[2].strip()
if taxid_1 in db:
tax_info = '\t'.join(db[taxid_1]) # Retrieve taxonomic information
f_out.write(f"{query_num}\t{acc_q_num}\t{taxid_1}\t{tax_info}\n")
print("All files processed.")和上面一样也需要给权限
chmod 755 NR_taxid_tax.py
# 批量
for i in NCBI_seqid_taxid_bacteria_*_result.txt
do
num=${i%%_result.txt}
/home/zhongpei/hard_disk_sda2/zhongpei/Software/my_script/NR_taxid_tax.py -i ${i} -db NCBI_Main.taxid.bacteria_new.txt -o ${num}_tax.txt
done
/home/zhongpei/hard_disk_sda2/zhongpei/Software/my_script/NR_taxid_tax.py -i NCBI_seqid_taxid_virus_result.txt -db NCBI_Main.taxid.virus_new.txt -o NCBI_seqid_taxid_virus_tax.txt
/home/zhongpei/hard_disk_sda2/zhongpei/Software/my_script/NR_taxid_tax.py -i NCBI_seqid_taxid_fungi_result.txt -db NCBI_Main.taxid.fungi_new.txt -o NCBI_seqid_taxid_fungi_tax.txt
/home/zhongpei/hard_disk_sda2/zhongpei/Software/my_script/NR_taxid_tax.py -i NCBI_seqid_taxid_archaea_result.txt -db NCBI_Main.taxid.archaea_new.txt -o NCBI_seqid_taxid_archaea_tax.txt
/home/zhongpei/hard_disk_sda2/zhongpei/Software/my_script/NR_taxid_tax.py -i NCBI_seqid_taxid_Eukaryotic_result.txt -db NCBI_Main.taxid.Eukaryotic_new.txt -o NCBI_seqid_taxid_Eukaryotic_tax.txt好了,可以交差了 !我宣布python是我们初学者的yyds
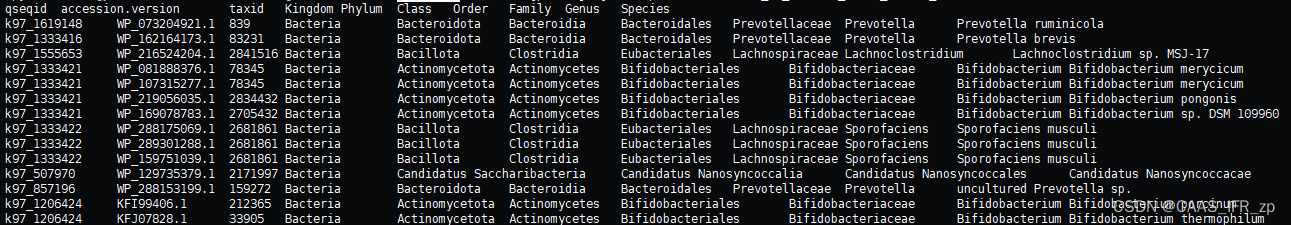
根据物种注释,计算宏基因组物种TPM相对丰度
这一步我也是想了一会,是按taxid进行group之后求和,还是按照物种注释的各种界门纲目科属聚类呢?最后还是决定使用界门纲目科属种,这样比较直觉,很多公司也是给的这个结果应该。
首先,我们需要一个TPM表格,是基于所有基因的。
这个可以参考之前的BWA-MEM2, Samtools: TPM定量-CSDN博客
其次,我们需要NR_taxid_tax.py这一步的结果,并且合并在一起
sed 's/>/\n>/g' *_tax.txt | sed '/^$/d' > ALL_tax.txt编写个代码,按照物种去求和TPM,同时可以处理界门纲目科属种水平
#! /usr/bin/env python
#########################################################
# TPM add up according to taxonomy file
# written by PeiZhong in IFR of CAAS
import argparse
import pandas as pd
parser = argparse.ArgumentParser(description='TPM count file combine')
parser.add_argument('--tpm', help='input tpm txt name')
parser.add_argument('--tax', help='input tax file name')
parser.add_argument('--out_prefix', help='output file prefix')
parser.add_argument('--level', '-l', type=str, help='Kingdom, Phylum, Class, Order, Family, Genus, Species, only use one in a time')
args = parser.parse_args()
tpm = args.tpm
tax = args.tax
out_prefix = args.out_prefix
level = args.level
ls_level = ["Kingdom","Phylum","Class","Order","Family","Genus","Species"]
if level not in ls_level:
raise ValueError("Invalid level specified. Use Kingdom, Phylum, Class, Order, Family, Genus, Species.")
# read TPM table
df = pd.read_csv(tpm, sep = "\t")
# read tax file
df2 = pd.read_csv(tax, sep="\t", header=0, dtype=str)
# delete some columns
df2.drop(columns=["accession.version", "taxid"], inplace=True)
# delete some headers
df2 = df2[~df2["qseqid"].str.contains("qseqid", na=False)]
# add new header
df2.columns = ["GeneID", "Kingdom", "Phylum", "Class", "Order", "Family", "Genus", "Species"]
# get level
if level == "Kingdom":
df2 = df2[["GeneID", "Kingdom"]]
else:
df2 = df2[["GeneID", "Kingdom", level]]
# merge df and df2 by column
merge_df = pd.merge(df, df2, on="GeneID", how="left") # merge df and df2 according to column1 (as key)
merge_df[level] = merge_df[level].fillna("Unassigned") # dont forget genes unmap by NR
# add TPM together according to bin_contig
kingdom_groups = merge_df.groupby("Kingdom")
if level == "Kingdom":
result_df = kingdom_groups.sum()
output_file = f"{out_prefix}_Kingdom.txt"
result_df.to_csv(output_file, sep="\t", float_format="%.4f")
print(f"Saved data to {output_file}")
else:
for kingdom, group in kingdom_groups:
group[level] = group[level].apply(
lambda x: f"Unassigned_{kingdom}" if x == "Unassigned" else x
)
result_df = group.groupby(level).sum()
output_file = f"{out_prefix}_{kingdom}_{level}.txt"
result_df.to_csv(output_file, sep="\t", float_format="%.4f")
print(f"Saved {kingdom} data to {output_file}")chmod 755 TPM_together_by_taxonomy.py
# Kingdom
TPM_together_by_taxonomy.py --tpm /home/zhongpei/diarrhoea/fan/metagenomic/assembly_result/goat_metagenomic_TPM.txt --tax ALL_tax.txt --out_prefix fan_all_cds_taxonomy -l Kingdom
# 门水平
TPM_together_by_taxonomy.py --tpm /home/zhongpei/diarrhoea/fan/metagenomic/assembly_result/goat_metagenomic_TPM.txt --tax ALL_tax.txt --out_prefix fan_all_cds_taxonomy -l Phylum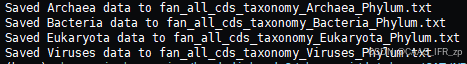
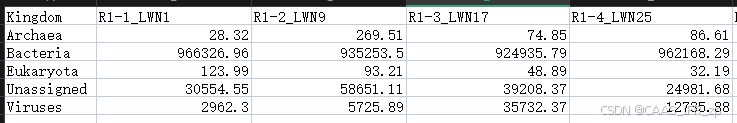
得出结果
大功告成!!


















 1万+
1万+

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









