






目录
- 研究者提出 EquiDTB 框架,将等变神经网络与 DFTB 方法结合,能以较低计算成本实现对大型有机分子的高精度模拟。
- 通过对阿比朵尔分子结构的策略性改造,研究者成功开发出抗新冠病毒效果显著增强且心脏安全风险大大降低的新候选药物。
- 在预测基因扰动后的表达变化时,简单的基线模型表现竟优于复杂的 scGPT 和 scFoundation,这揭示了当前基准数据集的不足。
- BioMiner 系统利用多模态 AI、领域模型和工具,实现了从海量文献中自动高效提取蛋白质 - 配体生物活性数据的创新。
- 研究者开发了一种结合可解释 AI 和增强采样的新方法,显著加速了 RNA 四聚体构象模拟,并揭示了其动态变化的关键驱动因素。
- DGCLCMI 是一种创新的深度图谱协作学习方法,能高效且准确地预测环状 RNA(circRNA)和微小 RNA(miRNA)之间的相互作用。
- 研究者推出 FoldScript 网络服务器,让分析和筛选 AI 生成的蛋白质模型变得简单高效。
- 研究发现,结合预训练 BERT 模型 (MolBERT) 和贝叶斯主动学习,能用更少的标注数据高效预测分子性质,加速药物设计。
- 研究者推出了 MultiFOLD2 和 ModFOLDdock2 这两个强大的网络服务器,用于精确预测蛋白质复合物结构并评估其质量。
- 研究者开发出一种 AI 新方法 (NISE),能从头设计出与特定小分子(如药物)紧密结合的蛋白质。
1. EquiDTB: 神经网络加速高精度分子模拟
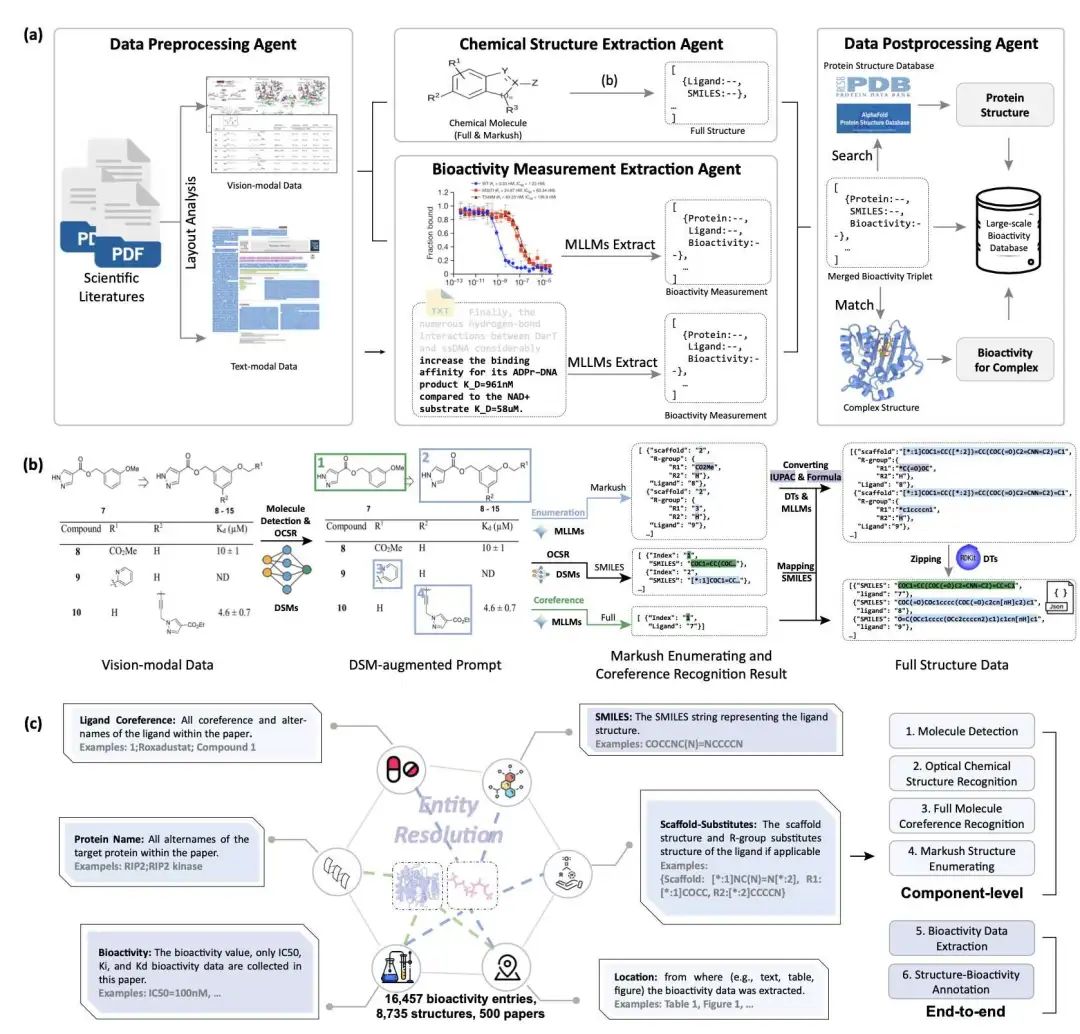
研究者提出了一个名为 EquiDTB 的新框架,巧妙地融合了密度泛函紧束缚 (DFTB) 方法与机器学习技术。这个混合方法的核心在于,它不再使用 DFTB 中标准的成对排斥势,而是引入了基于机器学习的 ∆TB 势。这种改变使得预测能量和原子间作用力更加可靠,尤其对于包含非共价相互作用或结构灵活的大型有机分子效果显著。
该方法采用了先进的 SE(3) 等变神经网络。这种网络能够更精准地捕捉分子内部和分子间的复杂相互作用,让模拟精度达到了 DFT-PBE0 的水准,同时还保持了较高的计算效率,足以应对大规模模拟的需求。因此,EquiDTB 成功克服了传统 DFTB 方法在处理特定体系,例如药物类化合物和生物分子时遇到的限制,特别是在处理非共价相互作用和分子柔性方面。
通过与标准的 DFTB3、GFN2-xTB 方法以及先前一些结合机器学习的尝试进行基准比较,EquiDTB 在计算相互作用能、原子受力以及振动频率等多个方面都展现出更低的误差和更优越的性能。它不仅能成功模拟像酪氨酸、扎普那斯特这类大型柔性分子和药物分子,还显示出良好的可扩展性和对训练集以外体系的迁移能力。
2. 阿比朵尔升级:抗新冠强效低毒
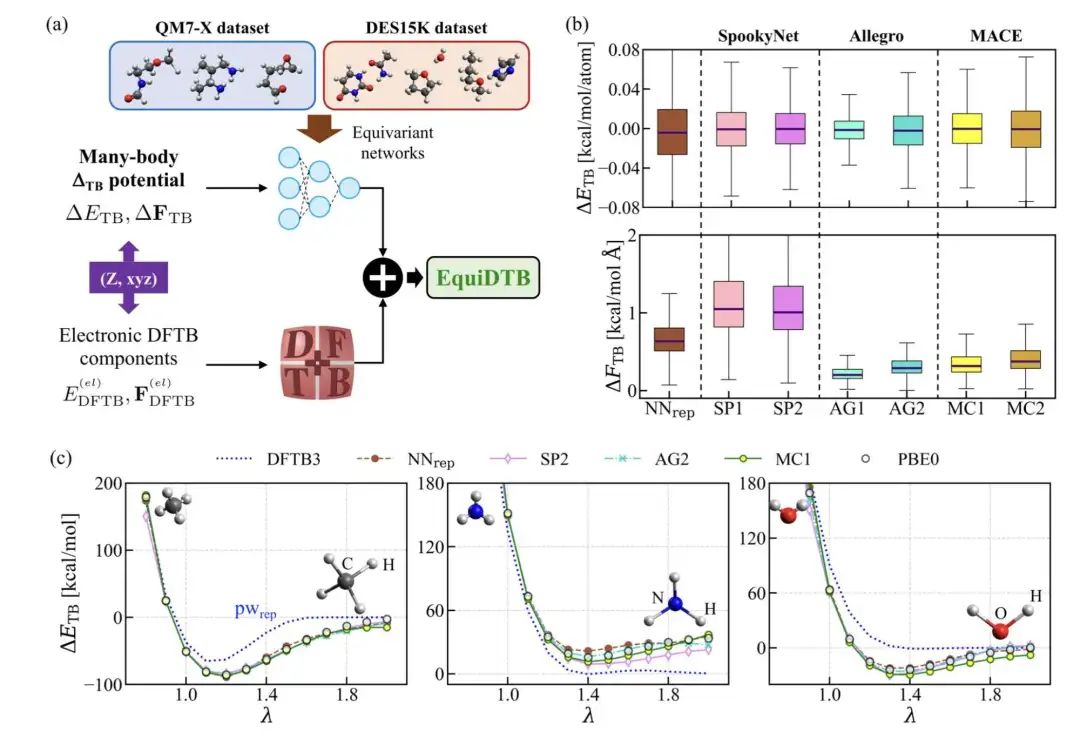
阿比朵尔(ARB,Umifenovir)是一种来自俄罗斯的广谱抗病毒药,但它并没有获得美国 FDA 的批准,主要是因为临床数据不够充分,而且有些潜在的毒性问题没弄清楚。不过,它独特的吲哚骨架结构,就像一个可以调整的平台,有六个可以修饰的地方,这给优化药物效果提供了可能。
这次研究的目标:一方面要提升阿比朵尔对抗新冠病毒(SARS-CoV-2)的“战斗力”,另一方面要确保药物更安全,特别是要降低对心脏的潜在风险(即 hERG 毒性)。研究者们动手修改了分子上的 N1、C2、C3 和 C4 这几个关键位置。
他们在感染了新冠病毒的 VERO E6 细胞上测试新分子的抗病毒效果。同时,药物在体内的吸收、分布、代谢、排泄(ADME)特性、动物实验中的表现(PK)以及心脏安全(hERG)评估,这些信息指导着优化的方向。
早期的尝试,比如修改 N1 和 C2 位置,就得到了化合物 10 和 14。它们的抗病毒效果(IC50 = 1.5 µM)已经比阿比朵尔(IC50 = 9.0 µM)强了不少。接着,进一步的精细调整产生了化合物 42(IC50 = 1.1 µM)和 56(IC50 = 0.24 µM),更棒的是,它们成功摆脱了心脏毒性的困扰(hERG 毒性 > 30 µM)。
随后,对 C3 位置的修改带来了更大的创新。研究者找到了几个“领头羊”化合物,比如 77、79 和 81。它们的抗病毒效果非常出色(IC50 在 0.67–0.7 µM 之间),并且同样没有了心脏毒性问题。
为了弄清楚这些新药是怎么起作用的,研究者们通过病毒进入抑制实验和免疫荧光技术进行了验证。结果证实,像化合物 77 这样的新分子,确实能有效地阻止新冠病毒进入细胞,并显著减少细胞内的病毒数量。这些发现表明,这些经过精心设计和优化的阿比朵尔类似物,非常有潜力成为下一阶段临床前研究的候选药物。
3. 细胞模型基准测试:简单方法意外胜出
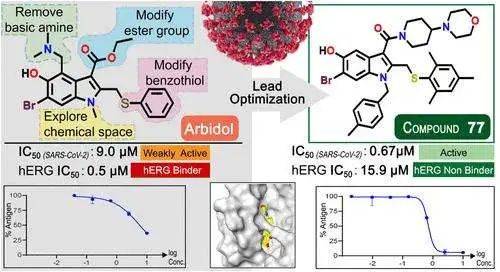
这项研究对两种基于 Transformer 的大型基础细胞模型——scGPT 和 scFoundation——进行了基准测试,目的是评估它们预测基因扰动后 RNA 测序(RNA-seq)表达谱的能力。这些模型预先在海量的未标记单细胞 RNA-seq 数据上进行了训练,理论上能利用学到的细胞嵌入信息来预测基因表达如何响应遗传干扰而变化。
然而,结果有点出人意料。研究者发现,一个非常简单的基线模型(直接使用训练数据的平均表达谱作为预测)在多个关键性能指标(比如皮尔逊相关系数)上的表现,竟然超过了这两个复杂的基础模型。这表明,尽管 scGPT 和 scFoundation 模型结构复杂、训练数据庞大,但在预测扰动效果这个具体任务上,它们并未展现出预期的优势。
为什么会这样呢?研究指出了当前常用基准数据集的一个大问题:它们包含的扰动类型不够丰富多样。这导致不同扰动实验下的细胞样本之间相似度很高,使得模型(尤其是复杂模型)很难学习到真正由特定扰动引发的独特变化。换句话说,数据集本身可能“太简单”,无法有效区分不同模型的真实能力。
更有意思的是,研究还发现,像随机森林这样相对传统的机器学习模型,如果给它加入一些具有生物学意义的特征(比如基因本体论 GO 注释),其预测效果也能优于 scGPT 和 scFoundation。这提示我们,在模型构建中,巧妙地融入领域知识(domain knowledge)可能比单纯追求模型的复杂度和数据量更为关键。
因此,这项工作强调,要想真正推动这个领域发展,需要开发出包含更多样扰动类型、覆盖更广泛生物学背景的数据集。同时,也需要更严格、更全面的基准测试策略,比如包含专门评估扰动特异性(PEX)和细胞类型特异性(CEX)任务的数据集,这样才能更准确地衡量和比较不同模型(特别是高级机器学习模型)在生物学预测任务中的实际应用价值。
4. BioMiner:AI 驱动的生物活性数据文献挖掘
从浩如烟海的科学文献中手动提取蛋白质与配体的生物活性数据,费时费力。为了解决这个问题,研究者开发了 BioMiner 系统。
这个系统混合使用了多种技术:既有多模态大语言模型(能看懂文字、图表),也有专门针对生物化学领域的模型和工具。这样一来,无论是普通的文字描述,还是表格、图片里的信息,甚至是像 Markush 结构这种复杂的化学表达方式,BioMiner 都能处理。
它的设计是模块化的,就像搭积木。研究者把整个复杂的提取过程拆分成了几个小任务,比如先解析文档,再提取化学结构,然后找出对应的生物活性测量值。这种方式的好处是不需要预先准备海量的、标注好的数据,也不用为每个小任务单独训练模型,省事不少。
为了检验 BioMiner 的效果,并且给后续研究提供一个标准,研究者还整理发布了一个叫 BIOVISTA 的数据集。这个数据集里包含了从 500 篇论文中精选出的 16457 条生物活性记录和 8735 个化学结构。在 BIOVISTA 上测试,BioMiner 提取生物活性三元组的 F1 分数达到 0.22,化学结构提取 F1 为 0.45,生物活性测量值提取 F1 为 0.53。更厉害的是它的速度,平均处理一篇论文只需要 14 秒。
BioMiner 不是纸上谈兵,它已经在实际场景中发挥了作用。比如,研究者用它处理了 11683 篇论文,建立了一个生物活性数据库,让相关模型的性能提升了 3.1%。在针对 NLRP3 靶点的研究中,它将高质量的生物活性数据量翻倍,使 QSAR 模型的性能大幅提高了 38.6%。它还能辅助研究人员进行蛋白质 - 配体结构标注,速度提升了 5 倍。系统还支持人工校验环节(HITL),专家可以检查确认提取出的数据,进一步保证了药物研发数据的准确性和效率。
当然,挑战依然存在。特别是要准确整合来自不同模态(文本、图像等)的复杂信息,以及完全理解那些高级的化学结构(比如 Markush),目前 F1 分数 0.22 的三元组提取结果也说明了这一点,还有提升空间。
5. AI 加速 RNA 构象模拟
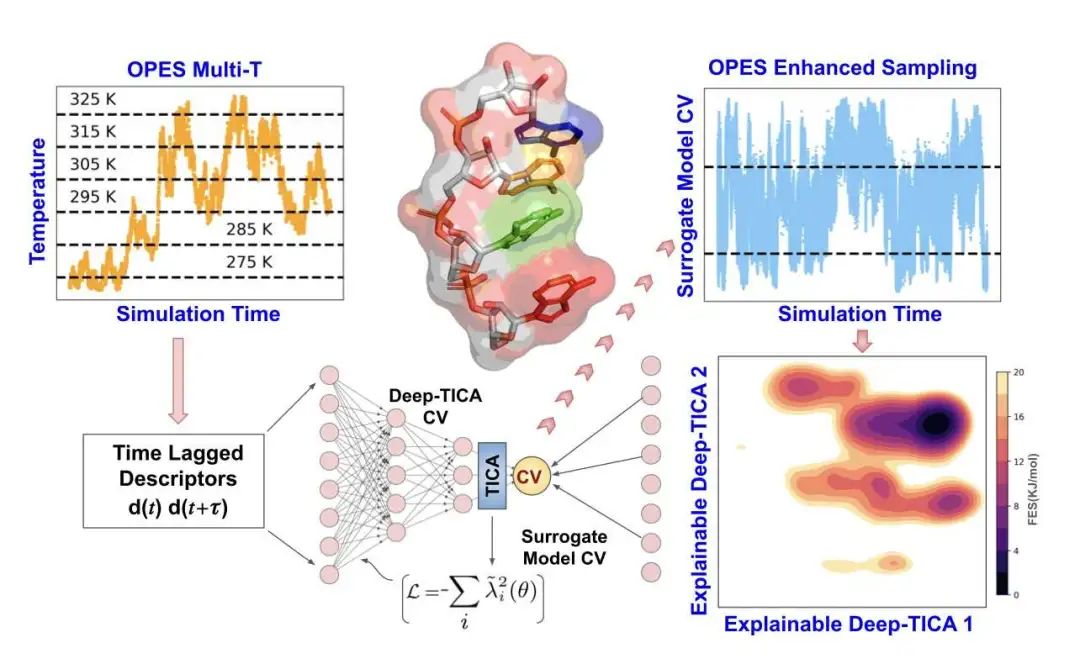
理解像 RNA 这样的小分子如何折叠和运动,对于揭示生命奥秘非常重要。但模拟这些过程通常很慢,计算成本很高。这项研究带来了一个新方法,把人工智能(特别是可解释 AI,简称 XAI)和一种叫做 OPES 的增强采样技术结合起来,专门用来研究 RNA 四聚体(就是四个核苷酸连在一起的小链,比如 AAAA、CCCC 和 UUUU)。
效果怎么样?快多了!
这种新方法的模拟速度,比老办法快了整整一百倍。它能更快地探索 RNA 可能呈现的各种形状,比如整齐堆叠的、相互嵌入的,或者是比较随意的卷曲结构。
这里用的 AI 不是一个“黑盒子”。通过可解释的深度学习模型(比如 Deep-TICA),研究者能弄清楚到底是 RNA 链上的哪些关键部位(主要是那些扭转角度)在控制着它从一种形状变成另一种形状的缓慢过程。这就好比不仅知道汽车在动,还知道是引擎的哪个部件在发力。这种理解对于深入了解 RNA 的工作方式非常有价值。
具体来说,研究者用了一种叫 Deep-TICA 的算法来识别 RNA 四聚体中最慢的变化部分,这有助于看清它们复杂的能量状态。他们还用了“代理模型”来简化 AI 的输出,把复杂的神经网络结果转换成更容易理解的分子特征(比如骨架和糖苷键的扭转角)的组合,这样既降低了计算负担,结果也更直观。
总的来说,这个新框架不仅大大加快了模拟速度,还能给出清晰的解释,告诉我们哪些特定的扭转角度控制着 RNA 形态间的转换。这不仅有助于改进现有的 RNA 力场模型,也为理解 RNA 的动态行为提供了新视角。而且,这个方法思路不只适用于 RNA,未来或许也能用来研究其他结构复杂、动态多变的生物大分子,比如一些本身结构就不太稳定的蛋白质。
6. DGCLCMI: 深度图谱预测 RNA 互作
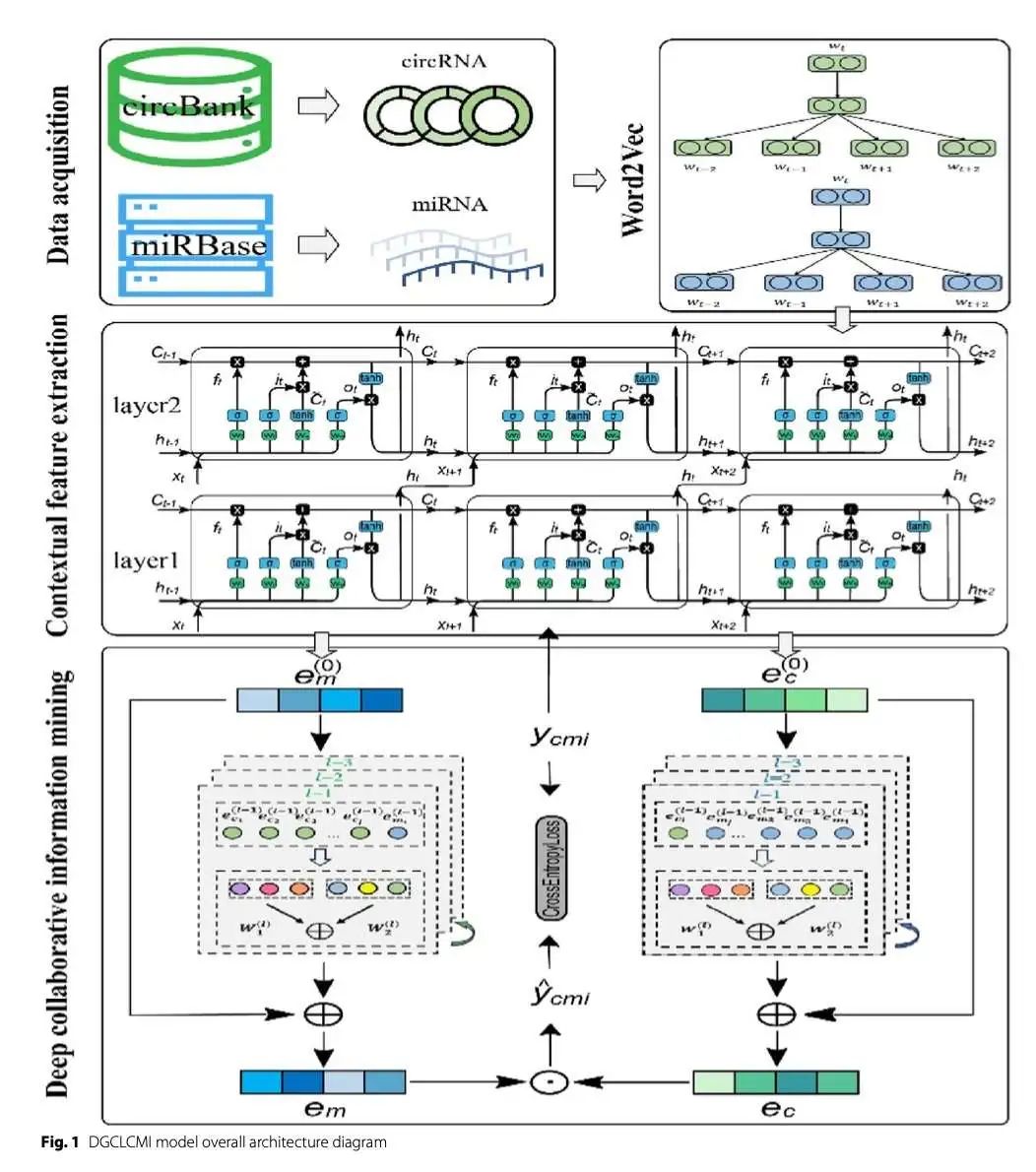
预测环状 RNA(circRNA)和微小 RNA(miRNA)如何相互作用,对于理解基因调控和疾病机制非常重要。为了解决这个难题,研究者开发了一种名为 DGCLCMI 的新方法。这是一种巧妙的深度学习技术,它把图谱学习和协作过滤结合起来,预测精度有了很大提升。
DGCLCMI 怎么工作呢?首先,它需要理解 RNA 序列。研究者使用了 Skip-gram 模型把 RNA 序列变成数字特征,然后用 LSTM 网络捕捉序列里的上下文信息。这样一来,模型就能更深刻地理解序列之间的关系。
这种方法的核心在于一个联合优化框架。它不是孤立地提取特征或挖掘互作信息,而是把这两步结合起来,一边提取序列特征,一边利用深度协作信号动态地优化这些特征。这就像是边学边用,让模型越来越聪明。
为了高效捕捉 circRNA 和 miRNA 之间复杂的依赖关系,DGCLCMI 采用了神经网络图谱协作过滤(NGCF)模型。这个模型能把互作的特点存储在各自的嵌入表示(embeddings)里,从而准确预测潜在的互作。
效果怎么样?在 CMI-20208、CMI-9589 和 CMI-9905 等多个公开数据集上,DGCLCMI 的表现都超过了现有方法,平均 AUC 值达到了惊人的 0.9600。这说明它区分真正互作和非互作的能力非常强。不仅如此,它在特异性、精确度和灵敏度方面也更好,AUC 和 AUPR 指标都有显著提高,证明了模型的稳健可靠。
理论好,实战如何?研究者进行了一个案例研究,用 DGCLCMI 来发现未知的 circRNA-miRNA 互作。结果令人鼓舞:在预测的 20 对潜在互作中,有 18 对得到了验证。这大大降低了后续生物实验验证的成本和时间。
相比那些静态的分类器,DGCLCMI 的一大优势是它的动态性和灵活性。因为它基于图谱的信息传播机制,可以实时调整,提取最有代表性的特征来进行高质量预测。
DGCLCMI 代表了计算生物信息学领域的一个重要进展。它为探索 circRNA-miRNA 互作提供了强大的新工具,有助于加深我们对疾病和基因表达调控网络的理解。
7. FoldScript:轻松搞定 AI 蛋白模型分析
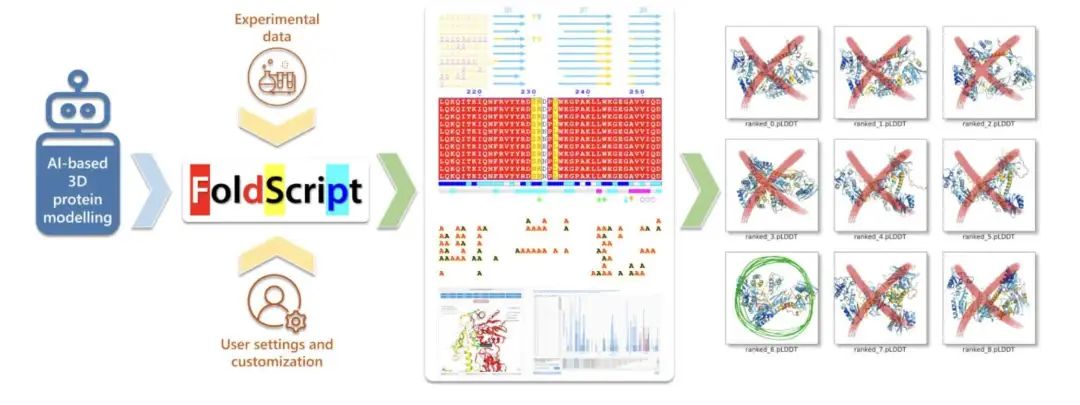
面对 AI 生成的大量蛋白质 3D 模型,如何高效筛选和分析成了新挑战。FoldScript 这个网页服务器应运而生,它让这项工作变得轻松不少。不同于只看评分最高的那个模型,FoldScript 能一口气整合多达 25 个模型的数据。这样一来,研究者就能更全面地了解蛋白质结构,找出最靠谱的预测结果。
这个工具支持像 AlphaFold3 和 RoseTTAFold 这样的主流 AI 预测器。用户可以方便地比较同一个蛋白质的不同模型,看看它们在结构上有什么差异。FoldScript 会生成清晰的图像,展示二级结构、模型可信度评分 (pLDDT),还有来自同源序列的比对信息。它的界面设计得很直观,不管是结构生物学专家还是不太懂行的人,都能比较容易上手。
一个特别有用的功能是接触分析模块。它可以识别蛋白质之间、蛋白质与配体或离子之间的分子接触点。这对于评估模型在模拟相互作用时的准确性非常有价值,尤其是在药物设计和分子生物学研究中评估寡聚体结构时,还可以结合实验知识来辅助筛选。
分析结果可以展示为平面图或者 3D 模型,方便快速进行大规模的结构评估。几分钟内就能搞定多个模型的初步评价。最棒的是,FoldScript 完全免费,也不需要注册登录,无论是学术界还是工业界的用户,都能无门槛使用这些强大的分析工具。开发者还计划未来让 FoldScript 支持 RNA 结构分析,加入更强的模型过滤功能,并处理更大的数据集,确保它能跟上 AI 蛋白质模型技术的发展步伐。
8. AI 药物设计:BERT+ 主动学习高效预测分子
对于新药研发,如何快速、准确地预测候选分子的性质,同时又尽量节省昂贵的实验资源?这项研究给出了一个新思路。
研究者将强大的预训练语言模型 BERT(在这里是专门用于分子的 MolBERT)和贝叶斯主动学习结合了起来。首先,MolBERT 在包含 126 万个化合物的大规模数据集上进行了预训练,这让它学会了深刻理解分子的“语言”,生成信息丰富的分子表示。有了好的分子表示,模型就能更准确地判断自己对哪些新分子“心里没底”,也就是所谓的不确定性估计。
然后,贝叶斯主动学习就派上用场了。它利用模型的不确定性估计,聪明地识别出那些对模型学习最有帮助、信息量最大的分子。这样,研究人员只需对这些“高价值”分子进行实验标记,而不是盲目地标记大量数据。结果如何?相比传统的主动学习方法,这种新方法达到同样预测效果所需的迭代次数减少了一半,大大节省了时间和成本。
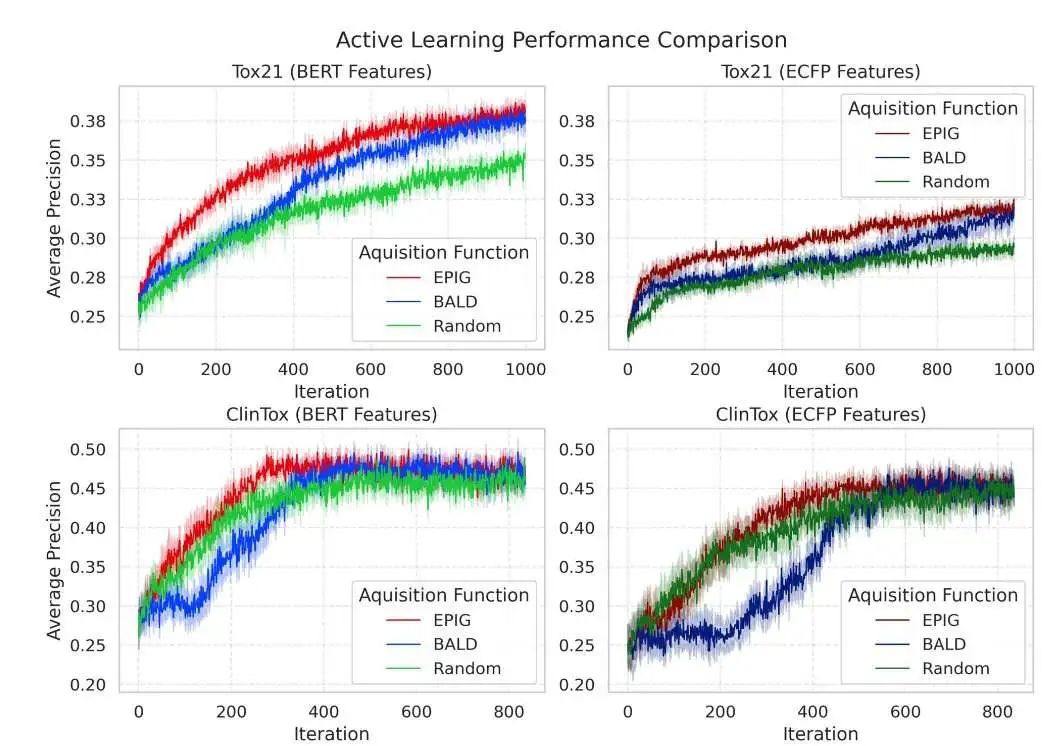
研究者在多个数据集上验证了这种方法的有效性,特别是在预测化合物毒性的 Tox21 和 ClinTox 数据集上。结果显示,基于 MolBERT 的主动学习方法不仅预测性能优于传统方法,而且模型校准得更好,选择样本的过程也收敛得更快。
研究中还比较了不同的主动学习策略(比如 EPIG 和 BALD 两种获取函数),发现 EPIG 配合 MolBERT 的表示,表现更稳定可靠。通过 UMAP 和 PCA 等可视化技术也能看到,MolBERT 产生的分子表示在特征空间里结构更清晰,这有助于主动学习更快、更准地找到有用的分子特征。
简单来说,高质量的分子表示(来自 MolBERT)极大地提升了不确定性估计的准确度,使得主动学习即使在初始数据很少的情况下,也能高效地挑出最有用的信息。这种数据高效的方法为加速早期药物筛选、特别是毒性预测,提供了一个强大的新工具,有望让新药研发流程更加高效和可扩展。
9. 预测蛋白质复合物:MultiFOLD2 与 ModFOLDdock2 服务器
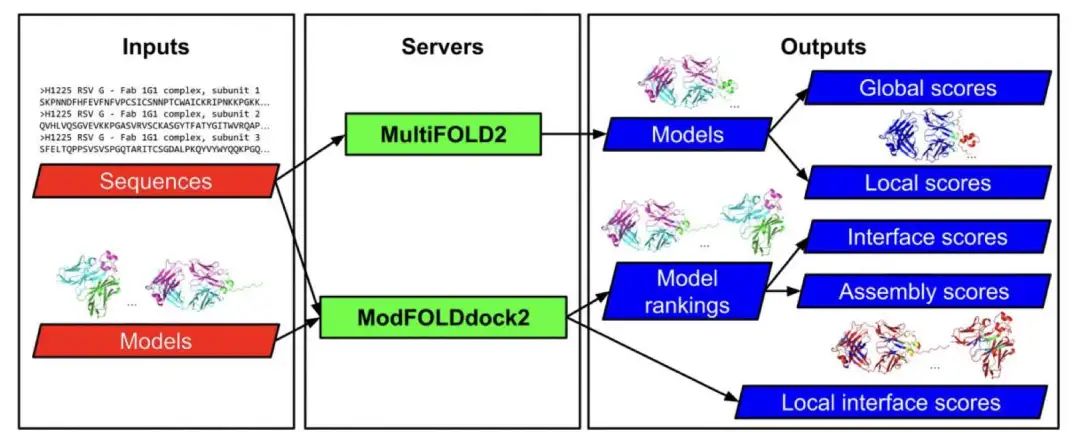
蛋白质很少单独行动,它们常常组成团队(即形成四级结构)来执行生物功能。弄清楚这些蛋白质团队如何组装起来对于理解生命过程至关重要,但这在技术上相当有挑战性。
现在,研究人员开发了两个新的在线工具来应对这个难题:MultiFOLD2 和 ModFOLDdock2。
MultiFOLD2 是一个预测工具。它的特别之处在于,即使研究者不确定一个蛋白质复合物由多少个单元组成(也就是化学计量未知),MultiFOLD2 也能尝试预测。它利用了当前顶尖的算法,比如 AlphaFold2-Multimer 和 RoseTTAFold2,来生成高质量的结构模型。在被誉为蛋白质结构预测领域“世界杯”的 CASP16 竞赛中,MultiFOLD2 在预测多聚体结构方面被评为表现最好的服务器之一,尤其擅长处理那些没有已知模板的复杂情况。它还运用了一些技术(如 dropout)来处理预测中的不确定性。
有了预测的模型,如何知道它有多可靠呢?这就是 ModFOLDdock2 发挥作用的地方。它像一个质量检查员,专门评估预测出的蛋白质复合物模型的准确性。它不仅能给出一个整体的质量评分,还能评估模型中蛋白质单元接触界面上每个氨基酸残基位置预测的准确度(局部质量)。ModFOLDdock2 采用一种混合多种信息的共识方法,在 CASP16 竞赛中评估模型质量方面同样表现突出。
更方便的是,这两个工具被整合在一起。用户可以在 MultiFOLD2 预测结构后,直接用 ModFOLDdock2 来评估结果的质量,整个过程在一个流畅的工作流程中完成。而且,这两个服务器都提供了简单易用的网页版,即使不是生物信息学专家也能轻松上手,利用这些前沿工具进行研究。
这些工具的出现,为研究蛋白质相互作用、药物发现以及理解疾病机理提供了有力的支持,有助于科学家们更深入地探索对生命科学至关重要的蛋白质四级结构。
10. AI 新器:零样本设计药物结合蛋白
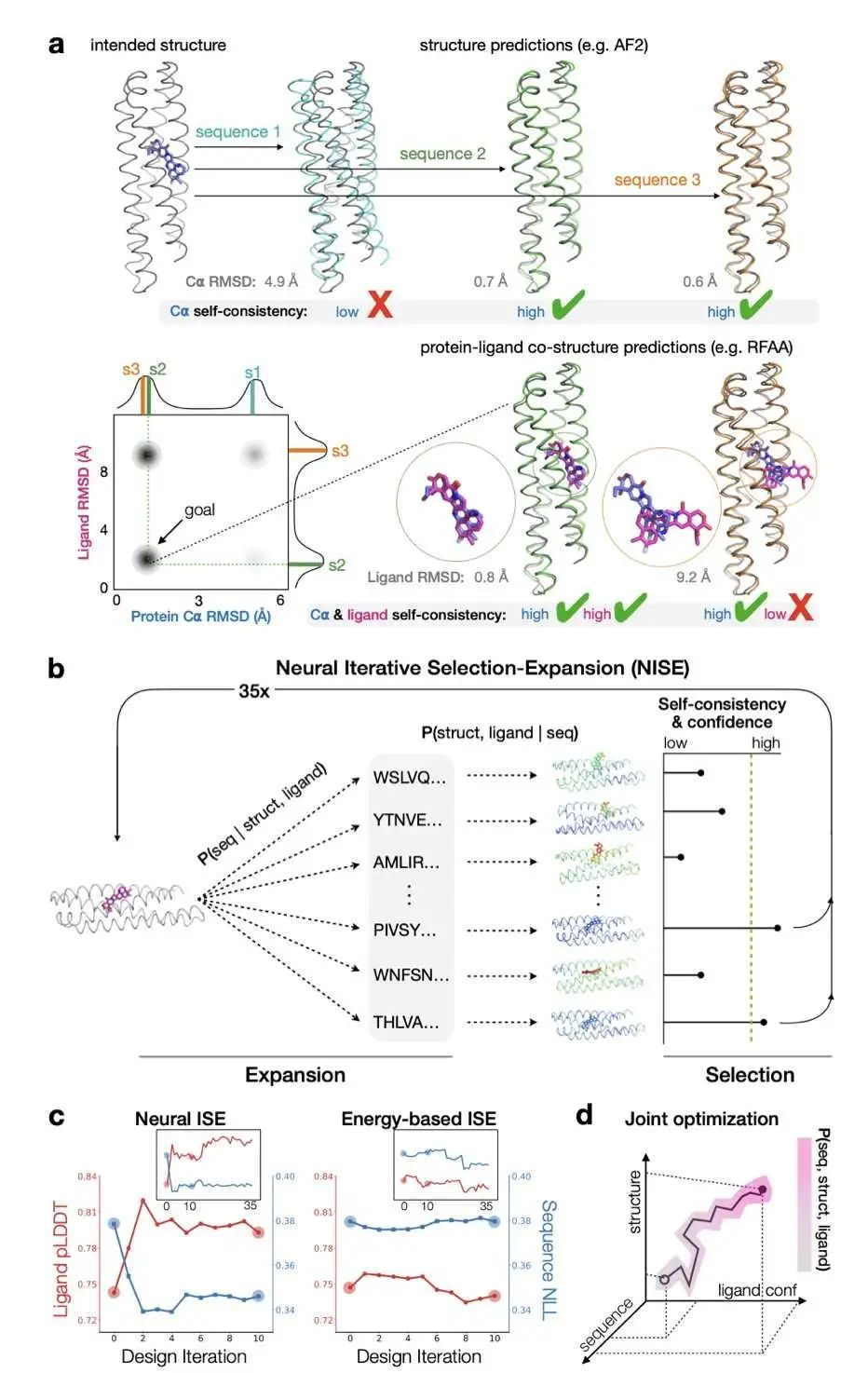
设计能与特定小分子(比如药物)结合的蛋白质,现在有了新的人工智能工具。研究者提出了一种叫 NISE 的新方法,它很聪明,能“零样本”设计出这类蛋白质,也就是不需要任何现有模板就能从头开始。
这个方法的核心是两个互相配合的神经网络:LASErMPNN 和 RoseTTAFold-All Atom (RFAA)。它们俩玩起了“你设计,我预测”的游戏。LASErMPNN 负责根据蛋白质和小分子的三维结构来设计蛋白质的氨基酸序列。然后,RFAA 接棒,根据设计出的序列来预测蛋白质和小分子结合后的三维结构。这个过程反复进行,不断优化,直到找到满意的设计。
为了验证这个方法的效果,研究者们挑战了一个实际任务:为抗癌药物 Exatecan 设计一个结合蛋白。Exatecan 是一种拓扑异构酶 I 抑制剂。结果相当不错!最好的设计能够以高达 120 nM 的解离常数 (Kd) 结合 Exatecan。这个亲和力比当前最好的方法还要强上 70 倍。
更厉害的是,研究者还引入了一种叫做“神经校对”的技术。LASErMPNN 不仅能设计,还能提出改进建议,指出哪些氨基酸突变可能让结合更紧密。仅仅采纳了两个预测的突变,Exatecan 结合蛋白的亲和力就飙升了 100 多倍,Kd 值达到了惊人的 1.2 nM!
Exatecan 在血液里不太稳定,容易水解失效,通常几个小时就没剩多少了。研究者测试了他们设计的蛋白质能否保护它。结果显示,表现最好的蛋白质 EPIC 能让 Exatecan 稳定好几天,这对于药物递送来说是个巨大的进步。
这项工作提供了一个通用的框架,可以用来设计各种小分子的结合蛋白。未来,它或许能在药物递送、解毒剂开发、生物传感器等领域大显身手。这表明,深度学习模型确实有能力自动化并加速设计过程,创造出高度特异性的蛋白质工具。当然,精确预测蛋白质和小分子如何相互作用,特别是小分子上那些关键的化学基团,仍然是个挑战。但随着结构预测技术的进步,这种设计方法的效果还会越来越好。
如何学习大模型 AI ?
由于新岗位的生产效率,要优于被取代岗位的生产效率,所以实际上整个社会的生产效率是提升的。
但是具体到个人,只能说是:
“最先掌握AI的人,将会比较晚掌握AI的人有竞争优势”。
这句话,放在计算机、互联网、移动互联网的开局时期,都是一样的道理。
我在一线互联网企业工作十余年里,指导过不少同行后辈。帮助很多人得到了学习和成长。
我意识到有很多经验和知识值得分享给大家,也可以通过我们的能力和经验解答大家在人工智能学习中的很多困惑,所以在工作繁忙的情况下还是坚持各种整理和分享。但苦于知识传播途径有限,很多互联网行业朋友无法获得正确的资料得到学习提升,故此将并将重要的AI大模型资料包括AI大模型入门学习思维导图、精品AI大模型学习书籍手册、视频教程、实战学习等录播视频免费分享出来。

第一阶段(10天):初阶应用
该阶段让大家对大模型 AI有一个最前沿的认识,对大模型 AI 的理解超过 95% 的人,可以在相关讨论时发表高级、不跟风、又接地气的见解,别人只会和 AI 聊天,而你能调教 AI,并能用代码将大模型和业务衔接。
- 大模型 AI 能干什么?
- 大模型是怎样获得「智能」的?
- 用好 AI 的核心心法
- 大模型应用业务架构
- 大模型应用技术架构
- 代码示例:向 GPT-3.5 灌入新知识
- 提示工程的意义和核心思想
- Prompt 典型构成
- 指令调优方法论
- 思维链和思维树
- Prompt 攻击和防范
- …
第二阶段(30天):高阶应用
该阶段我们正式进入大模型 AI 进阶实战学习,学会构造私有知识库,扩展 AI 的能力。快速开发一个完整的基于 agent 对话机器人。掌握功能最强的大模型开发框架,抓住最新的技术进展,适合 Python 和 JavaScript 程序员。
- 为什么要做 RAG
- 搭建一个简单的 ChatPDF
- 检索的基础概念
- 什么是向量表示(Embeddings)
- 向量数据库与向量检索
- 基于向量检索的 RAG
- 搭建 RAG 系统的扩展知识
- 混合检索与 RAG-Fusion 简介
- 向量模型本地部署
- …
第三阶段(30天):模型训练
恭喜你,如果学到这里,你基本可以找到一份大模型 AI相关的工作,自己也能训练 GPT 了!通过微调,训练自己的垂直大模型,能独立训练开源多模态大模型,掌握更多技术方案。
到此为止,大概2个月的时间。你已经成为了一名“AI小子”。那么你还想往下探索吗?
- 为什么要做 RAG
- 什么是模型
- 什么是模型训练
- 求解器 & 损失函数简介
- 小实验2:手写一个简单的神经网络并训练它
- 什么是训练/预训练/微调/轻量化微调
- Transformer结构简介
- 轻量化微调
- 实验数据集的构建
- …
第四阶段(20天):商业闭环
对全球大模型从性能、吞吐量、成本等方面有一定的认知,可以在云端和本地等多种环境下部署大模型,找到适合自己的项目/创业方向,做一名被 AI 武装的产品经理。
- 硬件选型
- 带你了解全球大模型
- 使用国产大模型服务
- 搭建 OpenAI 代理
- 热身:基于阿里云 PAI 部署 Stable Diffusion
- 在本地计算机运行大模型
- 大模型的私有化部署
- 基于 vLLM 部署大模型
- 案例:如何优雅地在阿里云私有部署开源大模型
- 部署一套开源 LLM 项目
- 内容安全
- 互联网信息服务算法备案
- …
学习是一个过程,只要学习就会有挑战。天道酬勤,你越努力,就会成为越优秀的自己。
如果你能在15天内完成所有的任务,那你堪称天才。然而,如果你能完成 60-70% 的内容,你就已经开始具备成为一名大模型 AI 的正确特征了。
这份完整版的大模型 AI 学习资料已经上传CSDN,朋友们如果需要可以微信扫描下方CSDN官方认证二维码免费领取【保证100%免费】



















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









