






RNA_seq下游分析
差异分析——利用R的DESeq2包
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("DESeq2")
library(DESeq2)
counts<-read.table("Male_count.csv",sep = ',',header = T,row.names = 1)
head(counts)
dim(counts)
mycounts<-counts[rowSums(counts) != 0,]
dim(mycounts)
#counts<-log2(counts+0.1)
#row.names一定要和数据的表头相一致
colDate<-data.frame(row.names=c("Water_1","Water_2","Water_3","Dss_1","Dss_2","Dss_3")
,condition=factor(c("Water","Water","Water","Dss","Dss","Dss"))
,levels=c("Water","Water","Water","Dss","Dss","Dss"))
head(colData)
dds<-DESeqDataSetFromMatrix(countData = mycounts,colData = colDate,design = ~condition)
dds<-DESeq(dds)
sizeFactors(dds)
res<-results(dds)
res
class(res)
res<-as.data.frame(res)
head(res)
library(dplyr)
res%>%
mutate(Sig = case_when(
log2FoldChange >= 0.58 & padj <= 0.05 ~ "UP",
log2FoldChange <= -0.58 & padj <= 0.05 ~ "DOWN",
TRUE ~ "Not_change"
)) -> res_1
table(res_1$Sig)
res_1<-cbind(rownames(res_1),res_1)
head(res_1)
colnames(res_1)<- c('gene',"baseMean","log2FoldChange","lfcSE","stat","pvalue","padj","Sig")
write.table(res_1,"Male_count_DEseq.csv",sep = ',',col.names = T,row.names = F,quote = FALSE)
Male_count.csv文件就是上篇推文得到的merged_counts.csv文件,只是把雄性样本的count挑了出来,命名为Male_count.csv
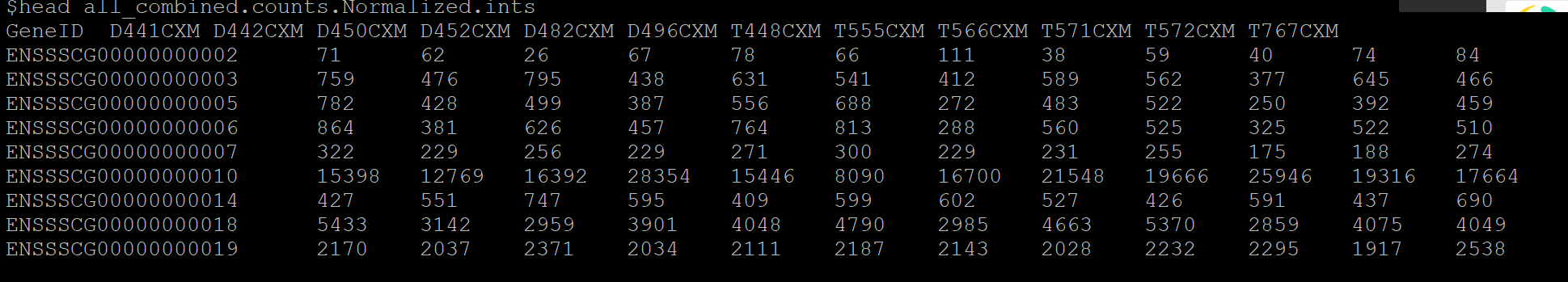
差异分析后,得到Male_count_DEseq.csv文件,但是还需要得到如下格式的表格all_combined.counts.Normalized.ints.DEseq2.txt.cut:
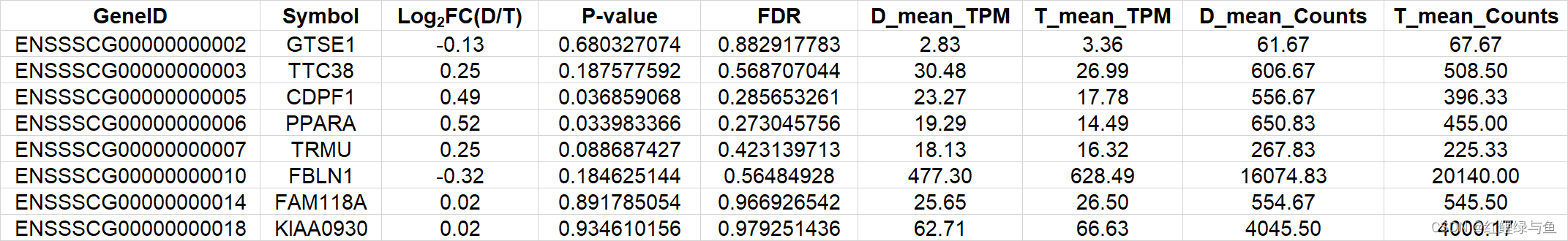 而现在的
而现在的Male_count_DEseq.csv文件格式如下:
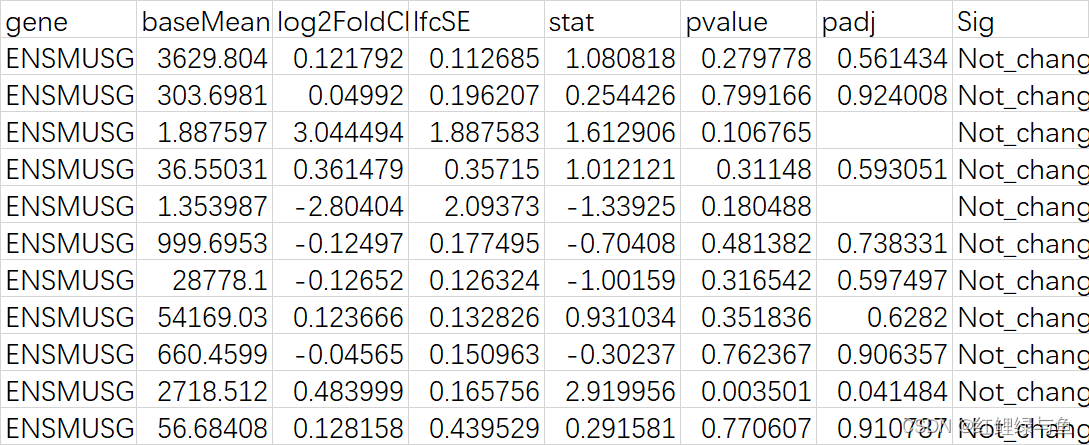 所以,还需要做如下处理:
所以,还需要做如下处理:
- 通过
gtf文件计算非冗余外显子的长度,从而计算TPM值,用于后续的可视化分析; - 将差异分析得到的
Male_count_DEseq.csv文件结合算出的TPM文件输出为all_combined.counts.Normalized.ints.DEseq2.txt.cut文件
计算非冗余外显子的长度
#!/usr/bin/perl
use warnings;
use strict;
my %hash;
open IN,"<","/workspace/home/pig/animal_geneticB/RNA_seq/reference/GRCm39/Mus_musculus.GRCm39.112.chr.PCGs.gtf" or die "Cannot open file /workspace/home/pig/animal_geneticB/RNA_seq/reference/GRCm39/Mus_musculus.GRCm39.112.chr.PCGs.gtf: $!";
open OUT, ">", "Mus_musculus.GRCm39.112.chr.PCGs.gtf.Efflens" or die "Can't open file Mus_musculus.GRCm39.112.chr.PCGs.gtf.Efflens: $!";
while(<IN>){
chomp;
next if /^#/;
my @a=split /\t/,$_;
if($a[2] eq 'exon'){
my $g=(split /;/,$a[8])[0];
$g=~s/gene_id "//;
$g=~s/"//;
foreach my $n($a[3]..$a[4]){
$hash{$g}{$n}=1;
}
}
}
close IN;
foreach my $g(sort keys%hash){
my $len=0;
foreach my $p(sort {$a<=>$b} keys%{$hash{$g}}){
$len++;
}
print OUT "$g\t$len\n";
}
close OUT;
命名为1_Calculate_Efflength.pl
运行perl代码
(base) [animal_geneticB@master-cli1-x86-agent1 5_DEseq]$ perl 1_Calculate_Efflength.pl
#得到以下数据
(base) [animal_geneticB@master-cli1-x86-agent1 5_DEseq]$ wc -l Mus_musculus.GRCm39.112.chr.PCGs.gtf.Efflens
21652 Mus_musculus.GRCm39.112.chr.PCGs.gtf.Efflens
(base) [animal_geneticB@master-cli1-x86-agent1 5_DEseq]$ head -n 10 Mus_musculus.GRCm39.112.chr.PCGs.gtf.Efflens
ENSMUSG00000000001 3262
ENSMUSG00000000003 902
ENSMUSG00000000028 3506
ENSMUSG00000000037 6397
ENSMUSG00000000049 1594
ENSMUSG00000000056 4806
ENSMUSG00000000058 3335
ENSMUSG00000000078 4391
ENSMUSG00000000085 7731
ENSMUSG00000000088 2101
计算TPM
#!/usr/bin/perl
use warnings;
use strict;
# 读取基因长度信息
my %LEN;
{
open my $IN, "<", "/workspace/home/pig/animal_geneticB/RNA_seq/5_DEseq/Mus_musculus.GRCm39.112.chr.PCGs.gtf.Efflens" or die "Cannot open gene length file: $!";
while (<$IN>) {
chomp;
my @a = split;
$LEN{$a[0]} = $a[1];
}
close $IN;
}
# 遍历命令行参数(所有filter文件)
foreach my $filter (@ARGV) {
open my $OUT, ">", "$filter.TPM" or die "Cannot open output file for $filter: $!";
my $cousum = 0;
# 计算总表达量 cousum
{
open my $IN, "<", "$filter" or die "Cannot open input file $filter: $!";
while (<$IN>) {
chomp;
my @a = split;
next unless exists $LEN{$a[0]};
my $r1 = ($a[1] / ($LEN{$a[0]} / 1000));
$cousum += $r1;
}
close $IN;
}
# 计算并打印每个基因的TPM值
{
open my $IN, "<", "$filter" or die "Cannot reopen input file $filter: $!";
while (<$IN>) {
chomp;
my @a = split;
next unless exists $LEN{$a[0]};
my $r1 = ($a[1] / ($LEN{$a[0]} / 1000));
my $tpm = keep2pos(1000000 * ($r1 / $cousum));
print $OUT "$_\t$tpm\n";
}
close $IN;
}
}
sub keep2pos{
my $inv = shift;
my $ouv;
if($inv < 0.01){
$ouv=0;
}else{
my @q=split /\./,$inv;
$ouv=$q[0] + 0.01*(substr($q[1],0,2));
}
return $ouv;
}
将上述脚本命名为2_Calculate_TPM.pl,在4_count文件夹中运行代码,得到.TPM文件,运行命令为:perl 2_Calculate_TPM.pl *.filter,可以查看文件的前十行:
##这是之前用含有非编码基因的gtf文件算出的TPM
(base) [animal_geneticB@master-cli1-x86-agent1 4_count]$ head -n 10 *.TPM
==> ERR10773103:Aligned.sortedByCoord.out.bam_htseq.counts.TPM <==
ENSMUSG00000000001 3186 98.5
ENSMUSG00000000003 0 0
ENSMUSG00000000028 243 6.99
ENSMUSG00000000031 0 0
ENSMUSG00000000037 27 0.42
ENSMUSG00000000049 2 0.12
ENSMUSG00000000056 790 16.57
ENSMUSG00000000058 24399 737.88
ENSMUSG00000000078 42899 985.35
ENSMUSG00000000085 584 7.61
##这是用只有编码基因的gtf文件算出的TPM
(base) [animal_geneticB@master-cli1-x86-agent1 4_count]$ head -n 10 *.TPM
==> ERR10773103:Aligned.sortedByCoord.out.bam_htseq.counts.filter.TPM <==
ENSMUSG00000000001 3186 101.24
ENSMUSG00000000003 0 0
ENSMUSG00000000028 243 7.18
ENSMUSG00000000037 27 0.43
ENSMUSG00000000049 2 0.13
ENSMUSG00000000056 790 17.03
ENSMUSG00000000058 24399 758.34
ENSMUSG00000000078 42899 1012.68
ENSMUSG00000000085 584 7.83
ENSMUSG00000000088 1678 82.78
从GTF文件中筛选出蛋白质编码基因的转录起始位点(TSS)
use strict;
my %CHR;
foreach my $c(1..18){
$CHR{$c}=1;
}
open IN,"<","/workspace/home/pig/animal_geneticB/RNA_seq/reference/GRCm39/Mus_musculus.GRCm39.112.chr.PCGs.gtf" or die;
open OUT, ">", "Mus_musculus.GRCm39.112.chr.PCGs.gtf.TSS" or die "Can't open file Mus_musculus.GRCm39.112.chr.PCGs.gtf.TSS: $!";
while(<IN>){
chomp;
next if /^#/;
my @a=split /\t/,$_;
#next unless (exists $CHR{$a[0]});
if($a[2] eq 'gene'){
if(/protein_coding/){
my $gid='';
my $gsm='Unknown';
my @b=split /;/,$a[8];
foreach my $f(@b){
$gid=$1 if ($f=~/gene_id \"(\S+)\"/);
$gsm=$1 if ($f=~/gene_name \"(\S+)\"/);
}
my $ginfo="$gid\_$gsm";
my $stp=($a[6] eq '+') ? $a[3] : $a[4];
my $enp=$stp+1;
print OUT "$a[0]\t$stp\t$enp\t$ginfo\n";
}
}
}
运行3_Pick_TSS_syobolID.pl,得到Mus_musculus.GRCm39.112.chr.PCGs.gtf.TSS文件,如下:
(base) [animal_geneticB@master-cli1-x86-agent1 5_DEseq]$ head -n 10 Mus_musculus.GRCm39.112.chr.PCGs.gtf.TSS
1 7159144 7159145 ENSMUSG00000051285_Pcmtd1
1 109910161 109910162 ENSMUSG00000026312_Cdh7
1 175708147 175708148 ENSMUSG00000039748_Exo1
1 175747895 175747896 ENSMUSG00000104158_Becn2
1 43866960 43866961 ENSMUSG00000057363_Uxs1
1 110905314 110905315 ENSMUSG00000047216_Cdh19
1 111792648 111792649 ENSMUSG00000038702_Dsel
1 176102878 176102879 ENSMUSG00000055214_Pld5
1 175806680 175806681 ENSMUSG00000078184_Rbm8a2
1 75427080 75427081 ENSMUSG00000033007_Asic4
输出表格
use strict;
my %MTPM;
open IN,"<","sample.list" or die;
open OUT, ">", "male_combined.counts.Normalized.ints.DEseq2.txt.cut.xls" or die "Can't open file male_combined.counts.Normalized.ints.DEseq2.txt.cut.xls: $!";
while(<IN>){
chomp;
next if /^Sample/;
my @a=split;
#open INN,"<","../2.Correlation/$a[0]\Aligned.out.bam.Counts.TPM" or die;
open INN,"<","/workspace/home/pig/animal_geneticB/RNA_seq/4_count/$a[0]:Aligned.sortedByCoord.out.bam_htseq.counts.filter.TPM" or die;
while(<INN>){
chomp;
my @b=split;
$MTPM{$a[1]}{$b[0]}+=$b[2];
}
close INN;
}
close IN;
my %AVTP;
my %MCOUNT;
open IN,"<","male_combined.counts.Normalized.ints" or die;
while(<IN>){
chomp;
next if /^GeneID/;
my @a=split;
my $v1=($a[1]+$a[2]+$a[3]);
my $v2=($a[4]+$a[5]+$a[6]);
#根据自己的数据生物学重复
$MCOUNT{'Wgroup'}{$a[0]}=$v1;
$MCOUNT{'Dgroup'}{$a[0]}=$v2;
}
close IN;
my %MAP;
open IN,"<","/workspace/home/pig/animal_geneticB/RNA_seq/5_DEseq/Mus_musculus.GRCm39.112.chr.PCGs.gtf.TSS" or die;
while(<IN>){
chomp;
my @a=split;
my @b=split /_/,$a[3];
$MAP{$b[0]}=$b[1];
}
close IN;
open IN,"<","male_combined.counts.Normalized.ints.DEseq2.txt.cut" or die;
while(<IN>){
chomp;
next if /^gene/;
my @a=split;
my $fc = 2 ** $a[1];
my $Dtpm=$MTPM{'Wgroup'}{$a[0]}/3;
my $Ttpm=$MTPM{'Dgroup'}{$a[0]}/3;
my $Dcou=$MCOUNT{'Wgroup'}{$a[0]}/3;
my $Tcou=$MCOUNT{'Dgroup'}{$a[0]}/3;
print OUT "$a[0]\t$MAP{$a[0]}\t$a[1]\t$fc\t$a[2]\t$a[3]\t$Dtpm\t$Ttpm\t$Dcou\t$Tcou\n";
}
##"3"是生物学重复的个数
运行4_Create_table.pl,得到male_combined.counts.Normalized.ints.DEseq2.txt.cut.xls文件,内容如下:
GeneID Symbol Log2FC(W/D) FC(W/D) P-value FDR W_mean_TPM D_mean_TPM W_mean_Counts D_mean_Counts
ENSMUSG00000000001 Gnai3 0.11512915 1.083071998 0.255436387 0.519218796 99.12 93.78666667 4685.666667 3101.333333
ENSMUSG00000000028 Cdc45 0.043080221 1.030311242 0.820703728 0.927784654 7.51 7.476666667 376 264.3333333
ENSMUSG00000000037 Scml2 0.355628115 1.279542542 0.302759047 0.568854664 0.54 0.436666667 51.33333333 28.66666667
ENSMUSG00000000049 Apoh -2.812191813 0.142378991 0.176770402 NA 0.02 0.126666667 0.333333333 2
ENSMUSG00000000056 Narf -0.132862349 0.912020179 0.440666351 0.692605683 16.94666667 19.03333333 1118 929
准备文件
sample.list文件
Sample Group
ERR10773113 Wgroup
ERR10773114 Wgroup
ERR10773121 Wgroup
ERR10773103 Dgroup
ERR10773104 Dgroup
ERR10773105 Dgroup
male_combined.counts.Normalized.ints文件
GeneID ERR10773113 ERR10773114 ERR10773121 ERR10773103 ERR10773104 ERR10773105
ENSMUSG00000000001 3392 7671 2994 3186 2823 3295
ENSMUSG00000000003 0 0 0 0 0 0
ENSMUSG00000000028 247 595 286 243 287 263
ENSMUSG00000000037 36 86 32 27 27 32
ENSMUSG00000000049 1 0 0 2 3 1
ENSMUSG00000000056 867 1587 900 790 961 1036
ENSMUSG00000000058 24460 49789 24544 24399 26613 29317
ENSMUSG00000000078 49299 121549 43026 42899 46210 49364
ENSMUSG00000000085 610 1365 468 584 529 692
male_combined.counts.Normalized.ints.DEseq2.txt.cut文件格式:
gene log2FoldChange pvalue padj
ENSMUSG00000000001 0.11512915 0.255436387 0.519218796
ENSMUSG00000000028 0.043080221 0.820703728 0.927784654
ENSMUSG00000000037 0.355628115 0.302759047 0.568854664
ENSMUSG00000000049 -2.812191813 0.176770402 NA
ENSMUSG00000000056 -0.132862349 0.440666351 0.692605683
ENSMUSG00000000058 -0.133991366 0.246420969 0.509966107
ENSMUSG00000000078 0.117121141 0.356336589 0.619214056
ENSMUSG00000000085 -0.052111137 0.719367375 0.876764984
ENSMUSG00000000088 0.476542556 0.003001567 0.035600623
ENSMUSG00000000093 0.122383369 0.786631677 0.913160197
ENSMUSG00000000094 0.305848441 0.898772916 NA
ENSMUSG00000000120 0.244867136 0.834132519 NA
注意:Sample和 all_combined.counts.Normalized.ints文件以及.TPM文件的表头一定要匹配上
sample.list文件标准格式:
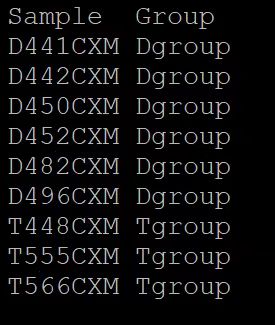
_combined.counts.Normalized.ints.DEseq2.cut文件标准格式:
表头并不匹配,应该是
gene_id、log2FoldChange、pvalue、padj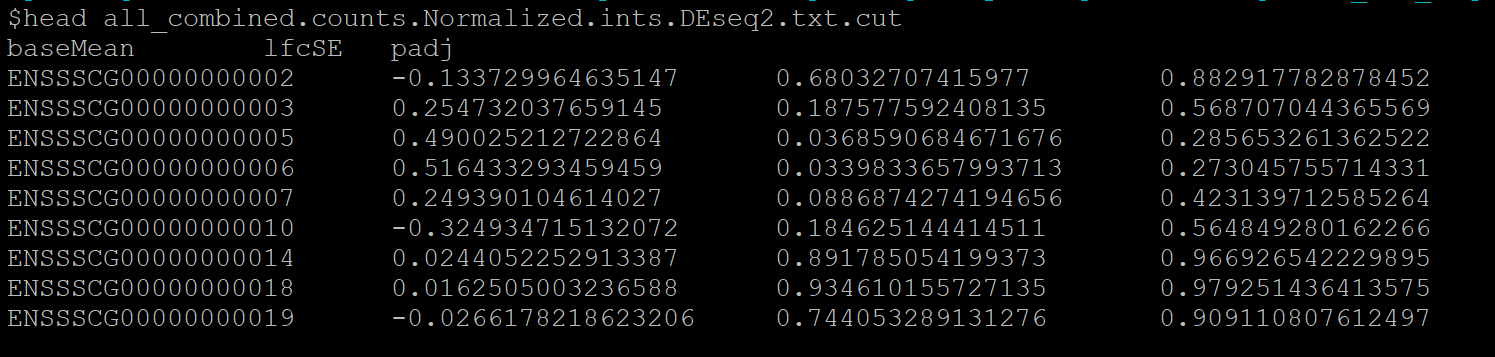
_combined.counts.Normalized.ints文件标准格式:
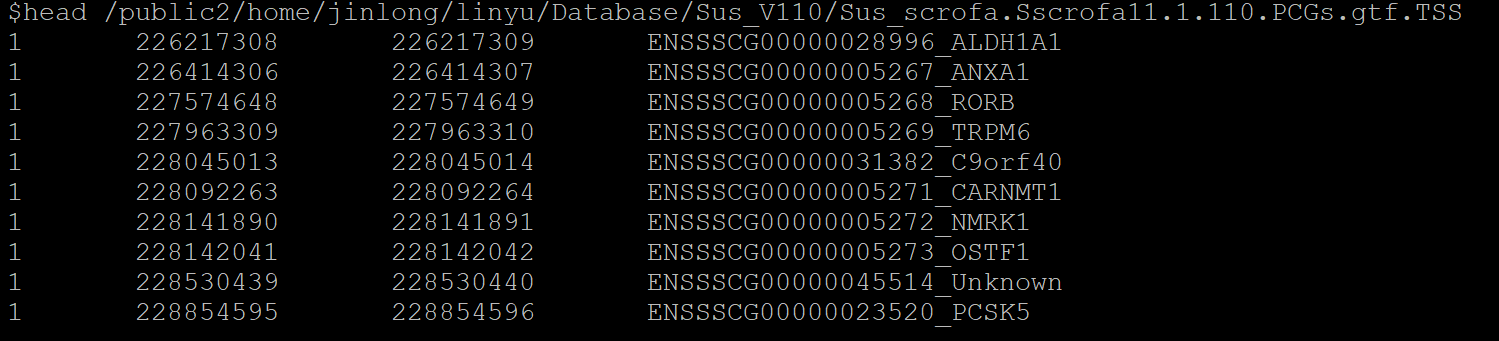
.gtf.TSS文件标准格式:















 1万+
1万+











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









