






应用场景:
- 基因预测和注释
- 基因结构分析
- 比较基因组学研究
- 转录本映射到参考基因组
主要用于进行基因组序列比对的命令。Exonerate 是一个灵活的基因组序列比对工具,特别适用于将 cDNA 序列比对到基因组序列上,以识别基因结构(外显子和内含子)。
安装:linux环境下或虚拟机可以直接conda安装
conda install bioconda::exonerate
conda install bioconda/label/cf201901::exonerate1.直接使用
最常用的无非就两个模型:
est2genome或protein2genome
区别在于是使用蛋白质还是CDS作为参照:
#使用CDS预测
exonerate \
--model est2genome \
--softmasktarget \
--showtargetgff \
--percent 70 \
--geneseed 200 \
--minintron 50 \
--maxintron 2000 \
--fsmmemory 1000 \
"ORF4_cds.fa" \
"oryza_orf4_cg.fa" \
> "oryza_orf4_cds.gff"
#使用蛋白预测
exonerate \
--model protein2genome \
--softmasktarget \
--showtargetgff \
--percent 70 \
--geneseed 200 \
--minintron 50 \
--maxintron 2000 \
--fsmmemory 1000 \
"ORF4_cds.fa" \
"oryza_orf4_cg.fa" \
> "oryza_orf4_cds.gff"
下面是对各参数的解释:
- `--model est2genome`: 使用 EST(表达序列标签)到基因组的比对模型,专门用于将 cDNA/mRNA 序列映射到基因组序列上,能够识别出外显子-内含子边界。
- `--softmasktarget`: 对目标序列进行软掩码处理,这样重复序列区域在比对过程中会被降低权重,但不会被完全忽略。
- `--showtargetgff`: 在 GFF(通用特征格式)输出中显示目标序列的信息。
这里可以展示目标序列在基因组中的相对位置信息- `--percent 70`: 设置序列相似性阈值为 70%,只有相似性达到这个阈值的比对结果才会被报告。
- `--geneseed 200`: 设置初始种子长度为 200bp,这是算法寻找潜在匹配区域的起始长度。
- `--minintron 50`: 设置最小内含子长度为 50bp,小于此长度的间隔不会被识别为内含子。
- `--maxintron 2000`: 设置最大内含子长度为 2000bp,大于此长度的间隔通常不被视为单个内含子。
- `--fsmmemory 1000`: 为有限状态机分配 1000MB 内存,控制程序运行时的内存使用。
- `"ORF4_cds.fa"`: 这里是目标CDS文件,fa格式即可。(如果使用整个基因或片段序列,相比blastn,exonerate会把目标序列更完整的识别,可能在处理同源目标片段存在大段插入缺失的时候处理效果更好。)
- `"oryza.genome.fa"`: 目标基因组fasta文件。
- `> "oryza_opt.gff"`: 结果文件,但并不是标准的gff。
这个命令的主要用途是将 ORF4 的 CDS(编码序列)比对到水稻基因组上,识别出该基因在基因组中的确切位置、外显子-内含子结构,结果以 GFF 格式输出,可用于后续的基因注释和分析。
相对于给定一个未知品种中的序列片段,其他软件直接预测最长的合理ORF,exonerate可以以CDS作为参照,尽可能的按照参照CDS中的剪接模式来预测这段未知片段中的CDS和包含此CDS的基因结构。要注意最大内含子长度的设定。
或者配合blastn一起使用,即先获得各物种中的基因片段,然后再通过exonerate预测基因结构与边界。
结果文件:
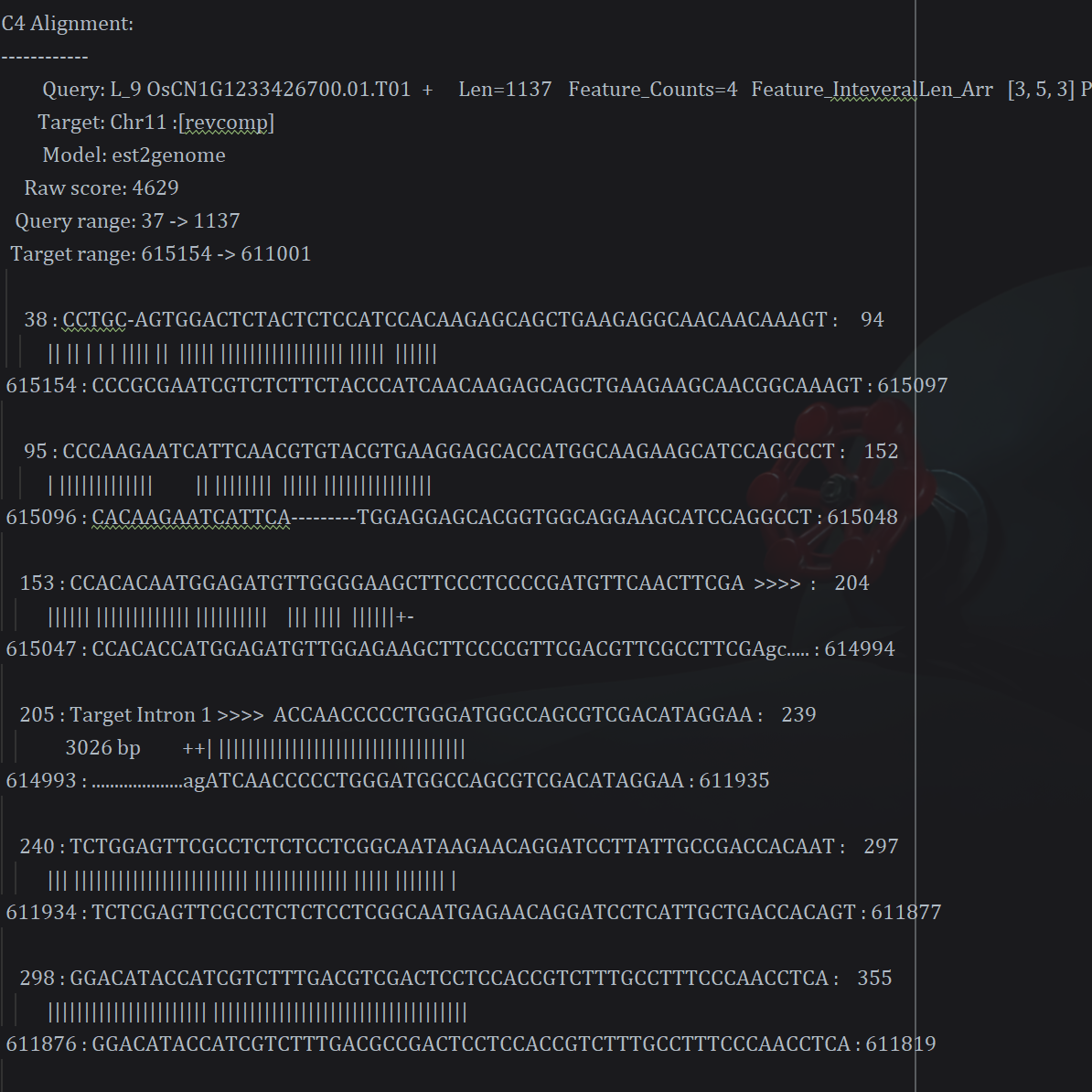
2.结果文件处理
目前也没有找到可以直接生成gff3或fasta匹配序列的方法。
所有很多后续功能的需求,使用一些简单的基础脚本就可以了
这里是输入CDS的情况下使用py正则提取目标匹配fasta序列的脚本:
import os
import re
def extract_():
with open(rf"input.gff") as f:
content = f.read()
alignments = []
for block in content.split('C4 Alignment:')[1:]:
name = re.findall(r'Target:(.+)\n', block)[0].strip()
seq = ''.join(re.findall(r'\|.+\n\s*\d+ : (.+) :', block))
seq = seq.replace(".", "").replace("-", '').replace("a", '').replace("t", '').replace("c", '').replace("g", '')
alignments.append(f">{name}\n{seq}")
# 调整顺序
alignments = alignments[::-1]
with open(f"output.fa", "w") as f:
f.write("\n".join(alignments))
#执行函数
extract_()
使用蛋白作为参照预测的结果文件处理脚本:
import re
amino_acids = {
'Ala': 'A', 'Arg': 'R', 'Asn': 'N', 'Asp': 'D',
'Cys': 'C', 'Gln': 'Q', 'Glu': 'E', 'Gly': 'G',
'His': 'H', 'Ile': 'I', 'Leu': 'L', 'Lys': 'K',
'Met': 'M', 'Phe': 'F', 'Pro': 'P', 'Ser': 'S',
'Thr': 'T', 'Trp': 'W', 'Tyr': 'Y', 'Val': 'V', '***': '*'
}
def convert_three_to_one(three_letter_seq):
# 将三字母序列按每三个字符分割
three_letter_codons = [three_letter_seq[i:i + 3] for i in range(0, len(three_letter_seq), 3)]
# 转换为单字母代码
one_letter_seq = ''.join(amino_acids.get(codon, 'X') for codon in three_letter_codons)
return one_letter_seq
# 处理序列
with open(r"input.gff") as f:
content = f.read().split("C4 Alignment:")[1:]
def re_program(segment):
# print(segment)
target = re.findall(r'Target: (.+)', segment)
seq_0 = re.findall(r'Target range.+?\n(.+?)\nvulgar:', segment, re.DOTALL)
seq_a = ""
for index, seq in enumerate(seq_0[0].split("\n")):
if (index - 3) % 5 == 0:
seq = seq.strip().rstrip().replace(" ", "")
seq_a += seq
seq_a = re.sub(r'{.+}', '', seq_a)
seq_a = re.sub(r'\+', '', seq_a)
seq_a = re.sub(r'\.', '', seq_a)
seq_a = re.sub(r'-', '', seq_a)
seq_a = re.sub(r'#', '', seq_a)
# seq_a = re.sub(r'-', '', seq_a)
# print(seq_a) # 正确的三位蛋白序列
one_letter_seq = convert_three_to_one(seq_a)
return target,one_letter_seq
with open("output.pr.fa", 'w') as f:
for segment in content:
result_0 = re_program(segment)
name,seq = result_0
f.write(f">{name[0]}\n{seq}\n")
-


















 1679
1679

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









