






S-RNase Evolution in Self-Incompatibility: Phylogenomic Insights into Synteny with Class I T2 RNase Genes
S-RNase在自交不亲和性中的进化:与Class I T2 RNase基因同源性的系统基因组学见解

摘要
S-RNases在多数开花植物的配子体自交不亲和(GSI)系统中起着至关重要的作用,它们作为花柱S决定因子发挥作用。尽管S-RNase基因在真双子叶植物中具有重要地位,但其同源基因组起源与进化轨迹仍不明确。本研究通过对130个被子植物基因组(涵盖35目56科)的T2型RNase基因进行大规模系统发育与微共线性网络分析,发现葫芦科物种的S-like RNase基因在系统发育树中与多个物种功能明确的S-RNase聚为一类。此外,葫芦科S-like RNase基因与Class Ⅰ T2 RNase基因显示出保守的基因组共线性。由此我们推断:已充分表征的S-RNase基因(属于Class III-A基因)与Class Ⅰ T2 RNase基因(位于重复基因组区块)可能共同起源于核心真双子叶植物共享的γ三倍化事件。研究还发现,S-RNase及S-like RNase基因在蔷薇科、茄科、芸香科等多个被子植物谱系中频繁发生谱系特异性基因转座,且伴随转座元件(TE)活性在相关基因附近显著增强。本研究首次阐明了真双子叶植物S-RNase基因的基因组起源与进化路径,深化了我们对基于S-RNase的GSI系统演化机制的理解。
背景
核糖核酸酶(RNases)在DNA复制、RNA代谢、植物防御及自交不亲和(SI)等多种细胞过程中发挥关键作用(Luhtala & Parker, 2010; MacIntosh, 2011)。其中,T2型RNase家族因具有两个保守活性位点而广泛存在于各类生物体中,发挥重要生物学功能(Irie, 1999)。在植物中,RNase T2基因家族通过基因重复和丢失发生显著扩张,导致不同物种间基因数量存在差异(MacIntosh et al., 2010)。基于系统发育分析及内含子数量,这些基因被划分为Class Ⅰ、Ⅱ、Ⅲ类(Igic and Kohn, 2001),其中S-RNase基因特属于Class Ⅲ类。尽管具有结构相似性,但各类基因在表达模式与功能上呈现显著差异:Class Ⅰ和Ⅱ类T2 RNase主要参与基因表达调控与抗菌防御反应,而S-RNase则是众多开花植物(被子植物)谱系中GSI系统的核心决定因子(Asquini et al., 2011; Franklin-Tong & Franklin, 2003; Hua et al., 2008; Liang et al., 2020; Ramanauskas & Igić, 2021; Takayama & Isogai, 2005)。
配子体自交不亲和(GSI)机制作为广泛存在的生殖隔离系统,在雌雄同体的开花植物中有效防止近交并促进异交(De Nettancourt, 2001)。目前学界确认的自交不亲和系统有四种类型:Ⅰ型GSI,发现于蔷薇科、茄科、车前科、芸香科、茜草科及仙人掌科植物中,通过基于S-RNase的机制发挥作用(Asquini et al., 2011; Franklin-Tong & Franklin, 2003; Hua et al., 2008; Liang et al., 2020; Ramanauskas & Igić, 2021; Takayama & Isogai, 2005)。Ⅱ型自交不亲和系统为孢子体自交不亲和(SSI),存在于十字花科植物中,依赖S位点受体激酶(SRK)与S位点富含半胱氨酸蛋白(SCR)发挥作用(Schopfer et al., 1999; Suzuki et al., 1999)。Ⅲ型GSI见于罂粟属植物,由罂粟柱头S蛋白(PrsS)和罂粟花粉S蛋白(PrpS)共同调控(Foote et al., 1994; Wheeler et al., 2009)。Ⅳ型自交不亲和系统见于报春花属植物,表征为孢子体异型花柱(Giacomo et al., 2022; Huu et al., 2020)。基于S-RNase的GSI系统在真双子叶植物中普遍存在,其遗传调控依赖于S位点,在自交不亲和反应中,S-RNase基因作为雌性决定因子,而S位点F-box(SLF)或S单倍型特异F-box(SFB)基因则作为雄性决定因子(Takayama & Isogai, 2005)。S-RNase与SLF/SFB基因的协同进化构成GSI系统的核心,并且被认为在双子叶植物中曾今发生过进化(Kubo et al., 2010; Steinbachs & Holsinger, 2002; Vieira et al., 2008)。针对T2型RNase基因——包括S-RNase基因在内——已在多个植物类群中有广泛研究,涵盖禾本科(Poaceae)、十字花科(Brassicaceae)、芸香科(Rutaceae)、豆科(Fabaceae)、蔷薇科(Rosaceae)及车前科(Plantaginaceae)物种(Azizkhani et al., 2021; Igic and Kohn, 2001; Liang et al., 2017; MacIntosh et al., 2010; Morimoto et al., 2015; Vieira et al., 2021; Zhu et al., 2023)。大量研究格外关注S-RNase的起源过程及其功能分化。这些研究采用了系统发育基因组学方法,整合现有基因组资源、比较基因组共线性关系,并推断了系统发育关系(Lv et al., 2022; Vieira et al., 2008; Zhao et al., 2022)。
尽管对真双子叶植物自交不亲和系统中S-RNase基因的演化有广泛的了解,但在T2 RNase家族中形成特定的S-RNase系统发育聚类的精确进化过程仍不明确。共线性信息在比较基因组学研究中具有关键价值,能为基因起源提供重要线索。微共线性网络分析作为系统发育研究的补充手段,可在基因组结构层面揭示基因亚家族间的深层位置关联(Zhao & Schranz, 2017; Zhao & Schranz, 2019)。本研究基于130种被子植物的基因组数据,对RNase T2基因家族开展全面共线性分析,重点解析S-RNase的基因组起源及其进化轨迹。
结果
被子植物RNase T2基因家族的系统发育分析
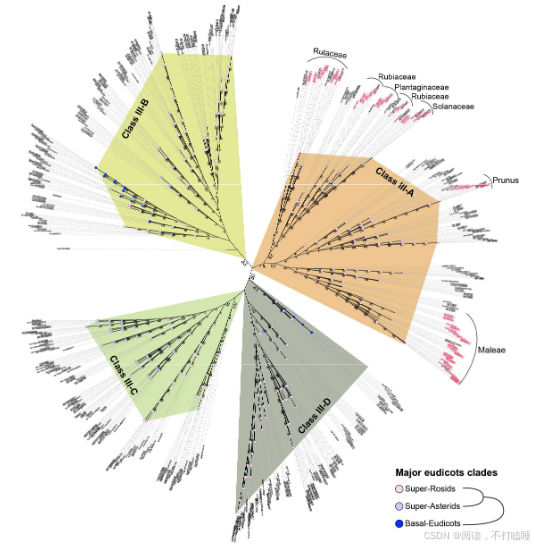
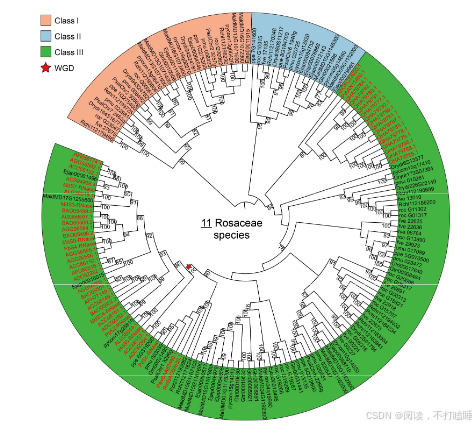
在本研究中,我们分析了涵盖被子植物各主要谱系的130个全基因组测序植物物种的RNase T2基因家族(附图S1,附表S1)。分析共鉴定出1366个T2 RNase基因(附表S2),该结果与以往研究结果具有高度一致性(附图S2)。基于所有鉴定到的T2 RNase基因(包含功能已知的S-RNase基因的蛋白质序列),构建了最大似然系统发育树(图1,附表S3)。该树主要划分为三个演化支:Class Ⅰ(Ⅰ类)、Class Ⅱ(Ⅱ类)及包含S-RNase的Class Ⅲ(Ⅲ类),符合当前系统发育分类框架(Igic & Kohn, 2001; Zhao等, 2022)。此外,我们进一步将这三类细分为七个主要亚类(图1,附图S3A)。
三类T2 RNase基因在被子植物各谱系中表现出不同的的进化模式。Class Ⅰ基因存在于所有主要被子植物类群中,包括真双子叶植物、木兰类、单子叶植物及ANA基部类群,表明其在整个被子植物进化历程中一直存在(图1,附图S3B)。Class Ⅱ基因分布于除ANA基部类群的核心被子植物中。相反,包含S-RNase的Class Ⅲ基因仅存在于真双子叶植物中,同时在十字花科(Brassicaceae)与菊科(Asteraceae)等谱系中观察到基因缺失或丢失现象(附图S3B)。
RNase T2基因家族拷贝数在被子植物类群间呈现显著差异,即使同科物种内部也存在很大波动,其范围从基部被子植物的3.3个拷贝到超菊类植物的12.9个拷贝(附图S3B)。值得注意的是,丛生四薮木(Durango root)和棉花等物种具有较多基因拷贝,而枣树(jujube)与秋海棠(begonia)等物种仅保留3个基因拷贝(附图S3B)。该基因家族的类型分布显示,ClassⅠ类(677个基因)与Class Ⅲ(470个基因)基因数量显著高于Class Ⅱ(219个基因),表明Class Ⅱ基因在植物进化过程中代表性相对有限,而Class Ⅰ基因经历了显著扩张(附图S3B)。我们的研究表明Class Ⅰ与Class Ⅱ基因数量间存在适度正相关(附图S4),而Class Ⅰ与Class Ⅲ、Class Ⅱ与Class Ⅲ基因间的相关性较弱(附图S4)。
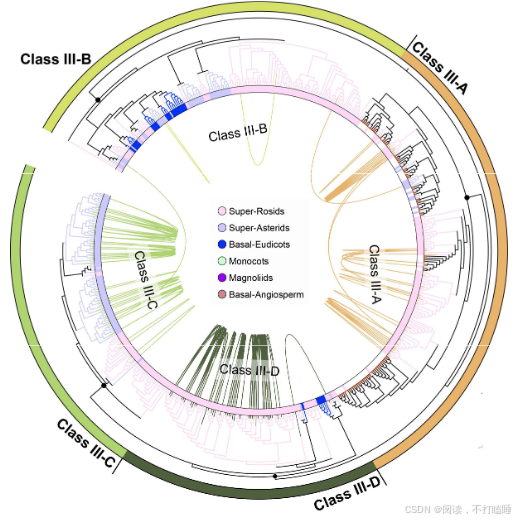
基于系统发育关系,我们将聚类树划分为七个主要亚类:Class Ⅰ-A、ClassⅠ-B、Class Ⅱ、Class Ⅲ-A(包含已报道的S-RNase基因)、Class Ⅲ-B、Class Ⅲ-C和Class Ⅲ-D(图1;附图S5A-B)。虽然没有发现Class Ⅲ-A基因特有的基序(附图S6),但蔷薇科S - RNase的基序组成与其他已报道的S-RNase(附图S6)差异很大。此外,我们还重点构建了包含不同物种(附图S7)已报道的S - RNase和S - like RNase的系统发育枝。为了验证已报道的代表性S - RNase的功能,我们对这些基因的表达情况以及它们与S位点F-box基因(SLFs)的基因组排列进行检测(附图S8-S9)。
在130种被子植物中,我们鉴定出五类基因复制事件(附图S10)。不同复制模式驱动了特定基因类群在植物谱系间的差异化扩张。全基因组复制(WGD)和串联重复(TD)是Class Ⅰ基因扩张的主要驱动力,特别是十字花科、白花菜科及棉属植物中表现显著。转座复制(TRD)主要促进了Class Ⅱ基因在蔷薇科和豆科等植物中的扩增。而包含S-RNase的Class Ⅲ基因演化主要依赖分散复制(DSD)机制(附图S10,附表S4-S5)。
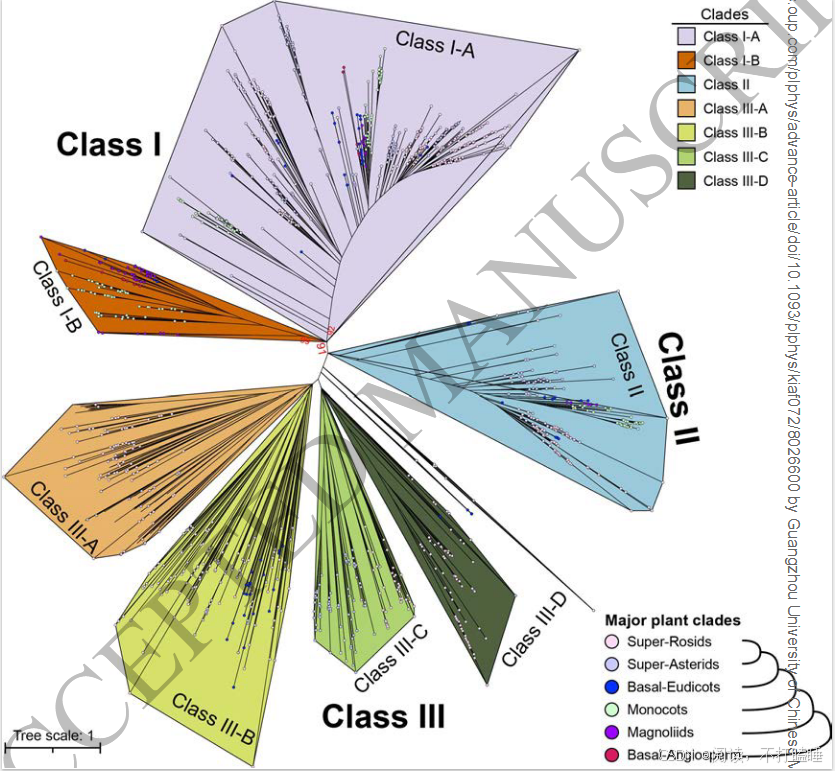
图1
附图S1:130种被子植物和11种蔷薇科植物基因组的系统发育关系。(A) 被子植物基因组 (B) 蔷薇科基因组。 已知的全基因组重复(WGD)和全基因组三倍(WGT)事件在系统发育树上显示。
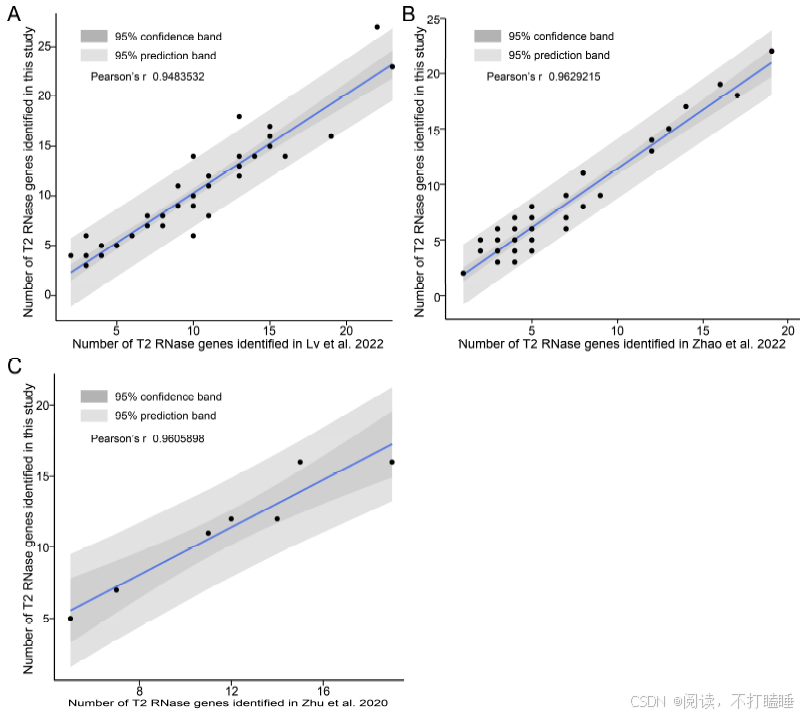
附图S2:不同研究中T2 RNase基因数量的比较分析。将本研究鉴定的T2 RNase基因数量与(A) Lv et al. (2022) (n = 50)、(B) Zhao et al. (2022) (n = 42)和(C) Zhu et al. (2020) (n = 7)报道的相应植物物种T2 RNase基因数量进行散点图比较。每个图中的黑线表示线性回归趋势,深灰色阴影表示95%置信区间,浅灰色阴影表示95%预测区间。
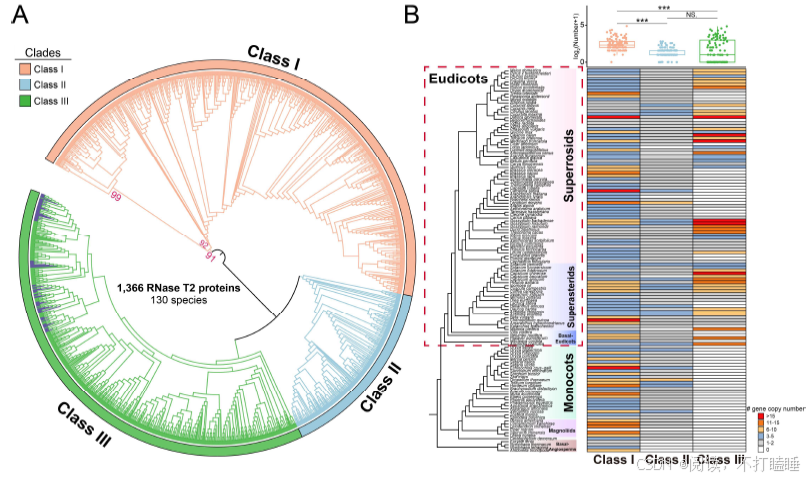
附图S3:130种被子植物RNase T2基因家族3大类的系统发育关系及基因计数 (A) 130种被子植物中RNase T2基因家族三大类的系统发育关系。已发表的功能性S-RNase基因用紫色突出显示。(B) Class Ⅰ、Class Ⅱ和Class Ⅲ基因拷贝数在植物个体间的分布。
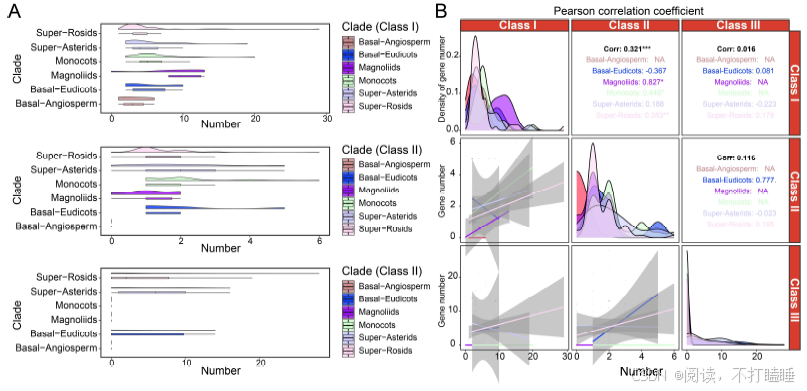
附图S4:不同枝上不同类的基因数据及相关性分析。(A) 图显示了super-rosids (n = 66), super-asterids (n = 23), monocots (n = 25), magnoliids (n = 6), basal-eudicots (n = 5), 和basal-angiosperms (n = 4) 基因组中T2 RNase基因的数量,以箱形图表示 (中线,中位数;盒限,上下四分位数;胡须,1.5×四分位间距;点,离群值)。(B) 不同进化支和分类中T2 RNase基因数量的Pearson相关系数(PCCs)分析Class Ⅰ(n = 130),Class Ⅱ(n = 120)和Class Ⅲ(n = 65)。相关系数显示在每个图的右上方。* p < 0.05;** p < 0.01;*** p < 0.001。
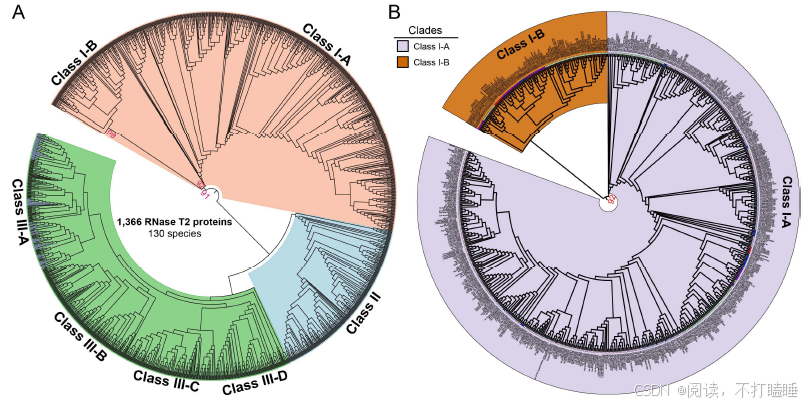
附图S5:130种被子植物RNase T2基因家族的系统发育关系 (A) 基于RNase T2基因家族全长蛋白序列,利用最大似然法(ML)构建的系统发育树。三种主要的种类都有不同的颜色,Class Ⅰ(橙色),Class Ⅱ(黄色)和Class Ⅲ(绿色)。(B) 从 (A) 和图1所示的完整T2 RNase系统发育中提取出来的Class Ⅰ分支。
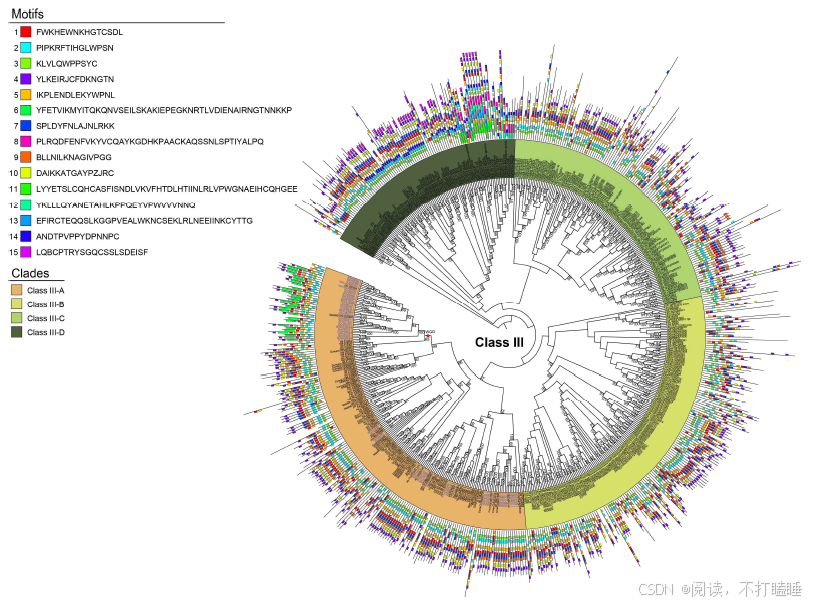
附图S6:130种被子植物Class Ⅲ T2 RNase基因的系统发育树和基序结构。
附图S7:Class Ⅲ T2 RNase基因的详细系统发育树。

附图S8:16个具有GSI系统的真双子叶植物雌蕊S-RNase基因的表达水平。
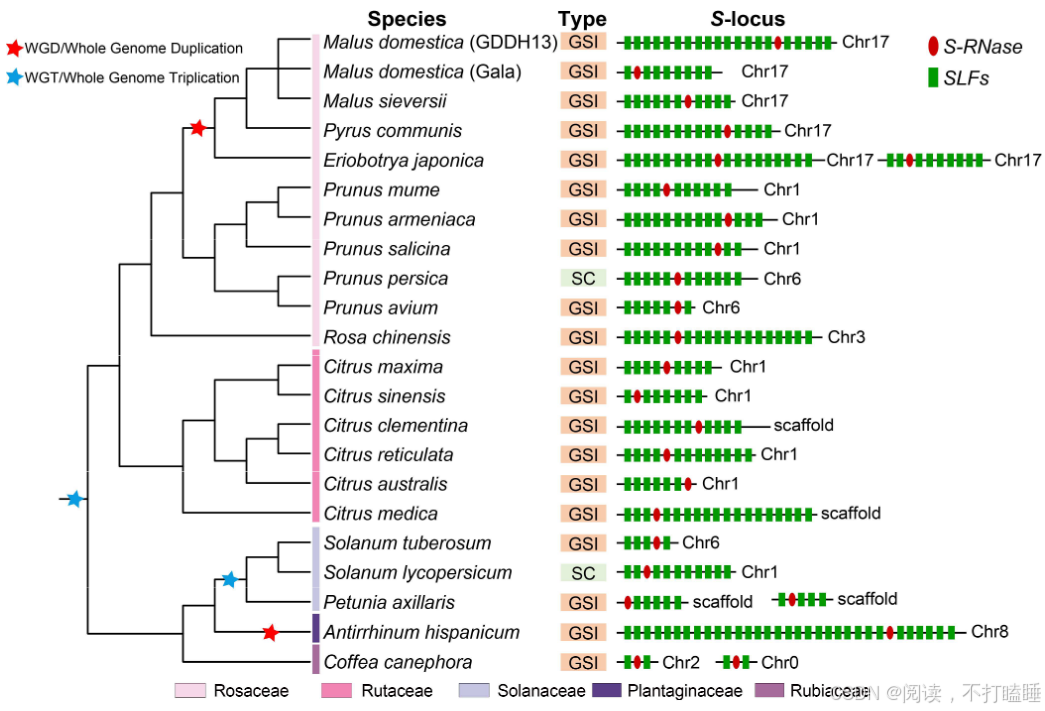
附图S9:5个科21种具有GSI系统的真双子叶植物的S位点、连锁S-RNases和SLFs基因组定位。
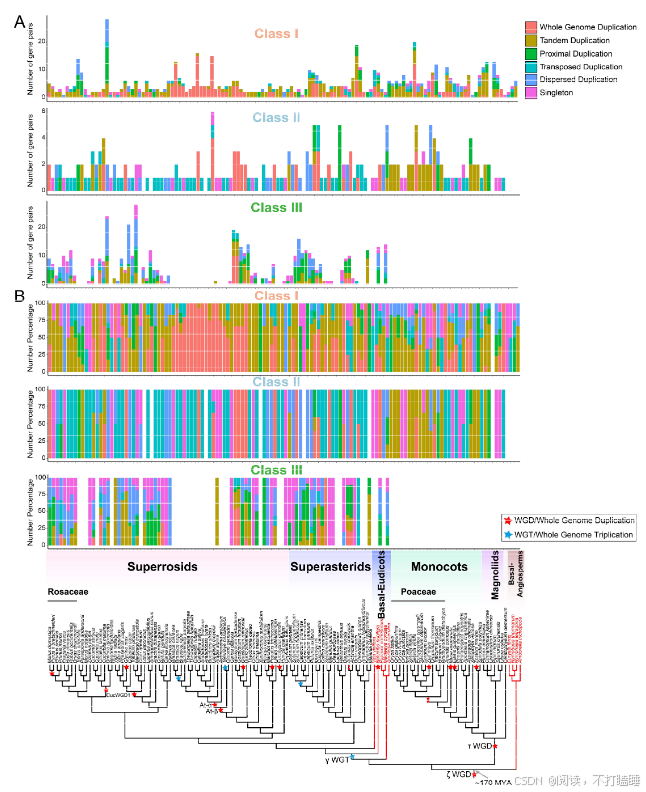
附图S10:已鉴定被子植物T2 RNase基因的基因复制模式。(A) 不同颜色的条形图表示每个物种在每种模式下复制的基因数量,包括全基因组复制(WGD)、串联复制(TD)、近端复制(PD)、转置复制(TRD)和分散复制(DSD)。(B) 各物种中不同类别基因复制的百分比。重复基因的优先顺序为:WGD >串联>近端>转置>分散。
基因组的保守性揭示了葫芦科Class Ⅲ-A S-like RNase基因与Class Ⅰ T2 RNase 之间共享祖先来源
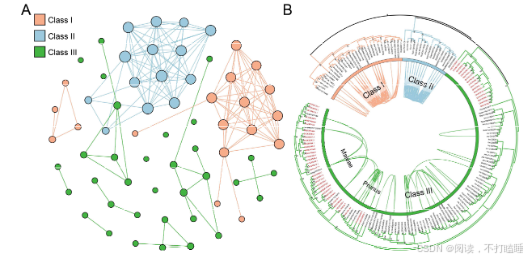
我们使用所有候选的T2 RNase基因从系统发育基因组微共线性数据库中提取了T2 RNase子网络。该网络由793个节点(T2 RNase基因)和19873条边(共线关系)(附图S11,附表S6)组成。利用Infomap聚类算法,我们识别出了67个共线性簇。Class Ⅰ基因相比其他类别的基因显示出更多的关联性,表明它们具有更强的共线性。相比之下,Class Ⅲ基因主要形成了小簇,表明它们在基因组背景下进化活跃。此外,Class Ⅲ基因显示出最低的基因共线性保留率(共线性网络中的基因数量相对于系统发育类群中基因数量的比例)和平均聚类系数(附图S12-S13)。
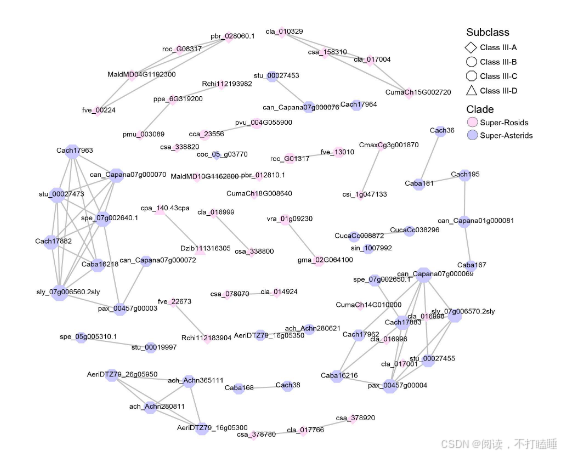
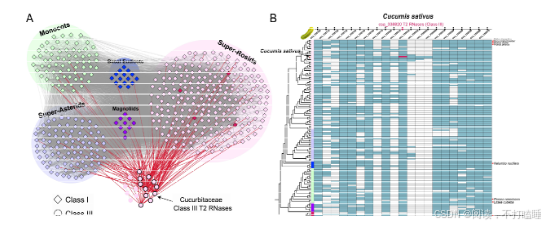
将T2 RNase基因的共线性关系映射到它们的系统发育树上,揭示了共线性与系统发育分类之间的普遍关联(图2A)。相比之下,在Class Ⅲ基因中观察到的共线性关联较少(图2A),但其保留的共线性仍支持四个亚类的分类(附图S14)。系统发育进化枝相对较低的支持值表明,该类中存在复杂的重复、丢失和序列分化历史(附图S15)。值得注意的是,我们观察到一些Class Ⅲ-A 类基因和Class Ⅰ T2 RNase基因之间存在显著的共线性保守性(图2A)。深入解析相关共线性簇发现,葫芦科物种(如黄瓜、西瓜、南瓜)的Class Ⅲ-A基因与其他被子植物Class Ⅰ基因存在特异性共线性关联(图2B,附图S16A)。
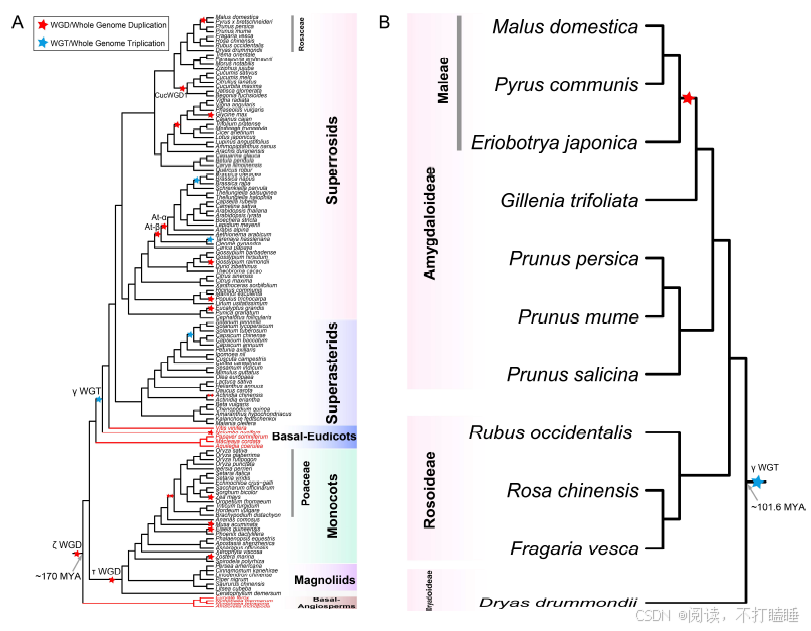
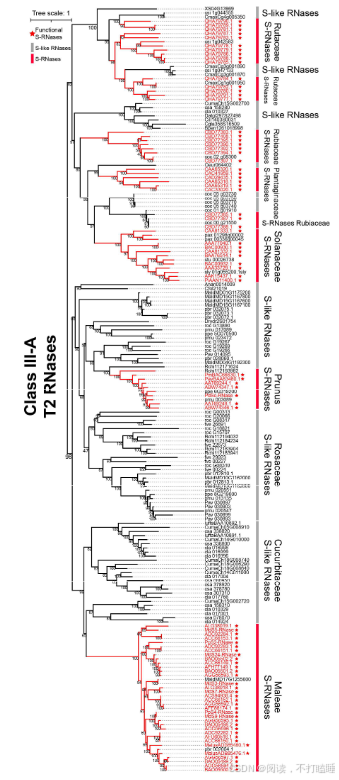
Class Ⅲ基因系统发育分析表明,所有功能明确的S-RNase基因均归属于Class Ⅲ-A分支(图3A,附图S7)。值得关注的是,葫芦科T2 RNase基因与已报道的苹果族(Maleae)S-RNase基因聚类关系最近,形成高支持率的进化分支(自展支持率BS=99%)(图3B,附图S7)。这一发现提示基于系统发育关系,可将葫芦科Class Ⅲ-A类T2 RNase基因鉴定为S-RNase的推定直系同源基因。然而,由于基于S- RNase的配子体自交不亲和性(GSI)并不适用于以单性花为主的葫芦科物种,我们将这些基因称为S-like RNase基因。
我们进一步研究了数据集中所有29种单性花物种的Class Ⅲ基因的系统发育分布(图3C)。结果表明,只有7个物种含有来自Class Ⅲ-A类进化枝的基因(图3C),且只有来自葫芦科物种的基因出现在共线性网络中。
我们描述了葫芦科(如黄瓜csa_338820)中Class Ⅲ-A类S-like RNase基因与基部被子植物、真双子叶植物等不同物种中相应的Class Ⅰ T2 RNase基因间的典型保守共线性区块(图3D, 附图S16B)。我们研究了这些葫芦科S-like RNase基因和共线性Class Ⅰ T2 RNase基因的表达模式。与来自苹果、梨和桃子的S-RNase基因相比,黄瓜Class Ⅲ-A类S-like RNase推定直系同源基因(如csa_338820)及其共线性Class Ⅰ基因(csa_285040)均呈现低表达模式(图3E)。这表明葫芦科Class Ⅲ-A类S-like RNase基因与Class Ⅰ T2 RNase基因间尚未发生显著功能分化。
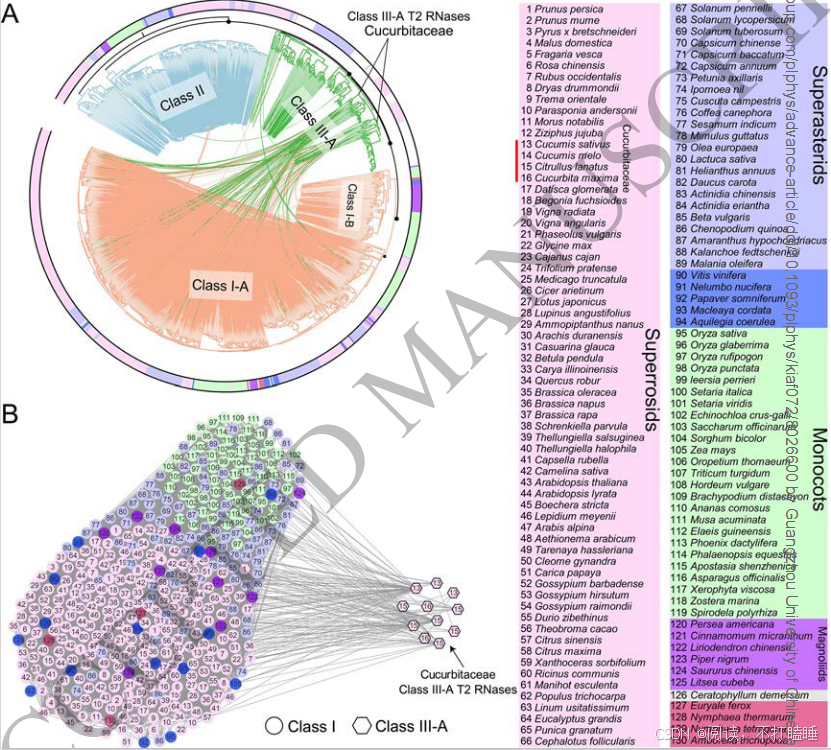
图2
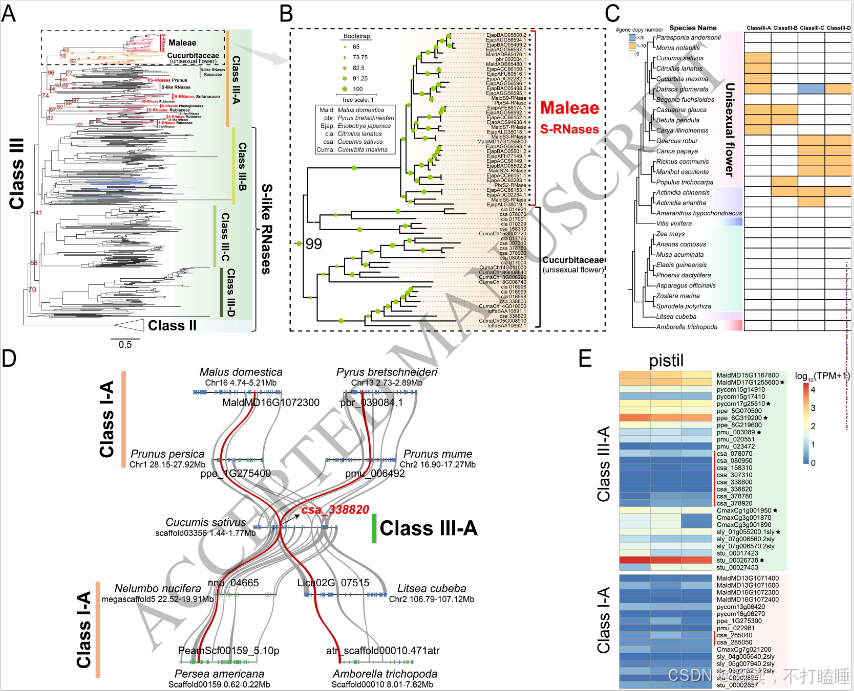
图3
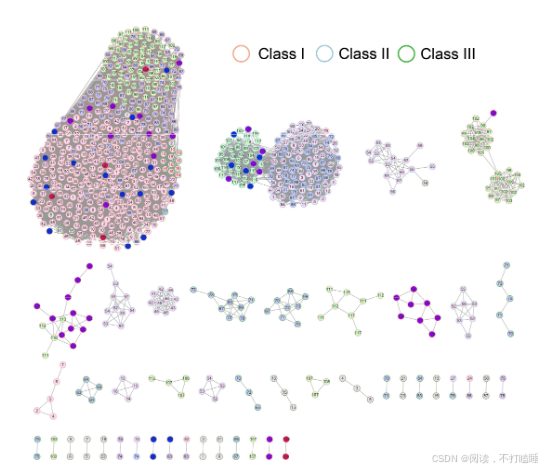
附图S11:130种被子植物中T2 RNase基因家族的同源染色体共线性关系网络图。
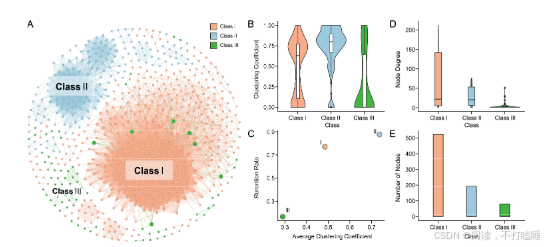
附图S12:三种RNase T2基因家族类别的共线性网络图和信息统计图。
附图S13:Class Ⅲ T2 RNase基因的共线性网络图。
附图S14:Class Ⅲ T2 RNase基因的最大似然基因树及相应的共线性关系。
附图S15: Class Ⅲ T2 RNase主要亚枝的无根树。
附图S16:葫芦科Class Ⅲ-A S-like RNase基因与被子植物Class Ⅰ S-like RNase基因的同源性和系统发育分析。(A) CIass Ⅲ和Class Ⅰ T2 RNase基因的共线性网络。(B) 黄瓜(Cucumis sativus)基因组中包含一个CIass Ⅲ T2 RNase基因 csa_338820(红色)的20个基因基因组背景。
黄瓜(Cucumis sativus)Class Ⅲ S-like RNase基因是γ全基因组三倍化事件中保留的重复基因产物
本研究采用系统发育分析与祖先共线性区块重建技术,解析葫芦科CIass Ⅲ-A S-like RNase基因与CIass Ⅰ T2 RNase基因的起源。同样,葫芦科T2 RNase基因可划分为三类,并发现CIass Ⅲ基因与CIass Ⅰ T2 RNase基因存在共线性区块,如黄瓜的csa_338820/csa_285040基因对之间,西瓜的cla_016996/cla_010187基因对之间(图4A)。
通过分析黄瓜(Cucumis sativus)、西瓜(Citrullus lanatus)及南瓜(Cucurbita maxima)三种葫芦科植物基因组内共线性基因对的同义核苷酸替换率(Ks)分布,揭示了两个多倍化事件的一致模式(图4B)。具体而言,Ks~1.5的峰值对应已报道的葫芦科共有四倍化事件(CucWGD1)(图4B)(Ma et al., 2022),而Ks~2.0的峰对应于核心葫芦共有的全基因组三倍化事件(γ)事件(Jiao et al., 2012)。结合我们所确定的"CIass Ⅲ-A S-like RNase与CIass Ⅰ"共线性基因对Ks值(如csa_338820/csa_285040:1.99;cla_016996/cla_010187:2.21),可以推断葫芦科CIass Ⅲ-A S-like RNase基因起源于γ三倍化事件。
此外,通过将黄瓜(Cucumis sativus)基因组定位至祖先核心真双子叶植物核型(Ancestral Core Eudicots Karyotype, ACEK)时,黄瓜基因csa_338820(位于3号染色体上)和CIass Ⅰ T2 RNase基因csa_285040(位于5号染色体上)分别对应于ACEK的3号染色体和17号染色体(图4C)。这一结果再次表明它们起源于双子叶植物 γ三倍化事件。
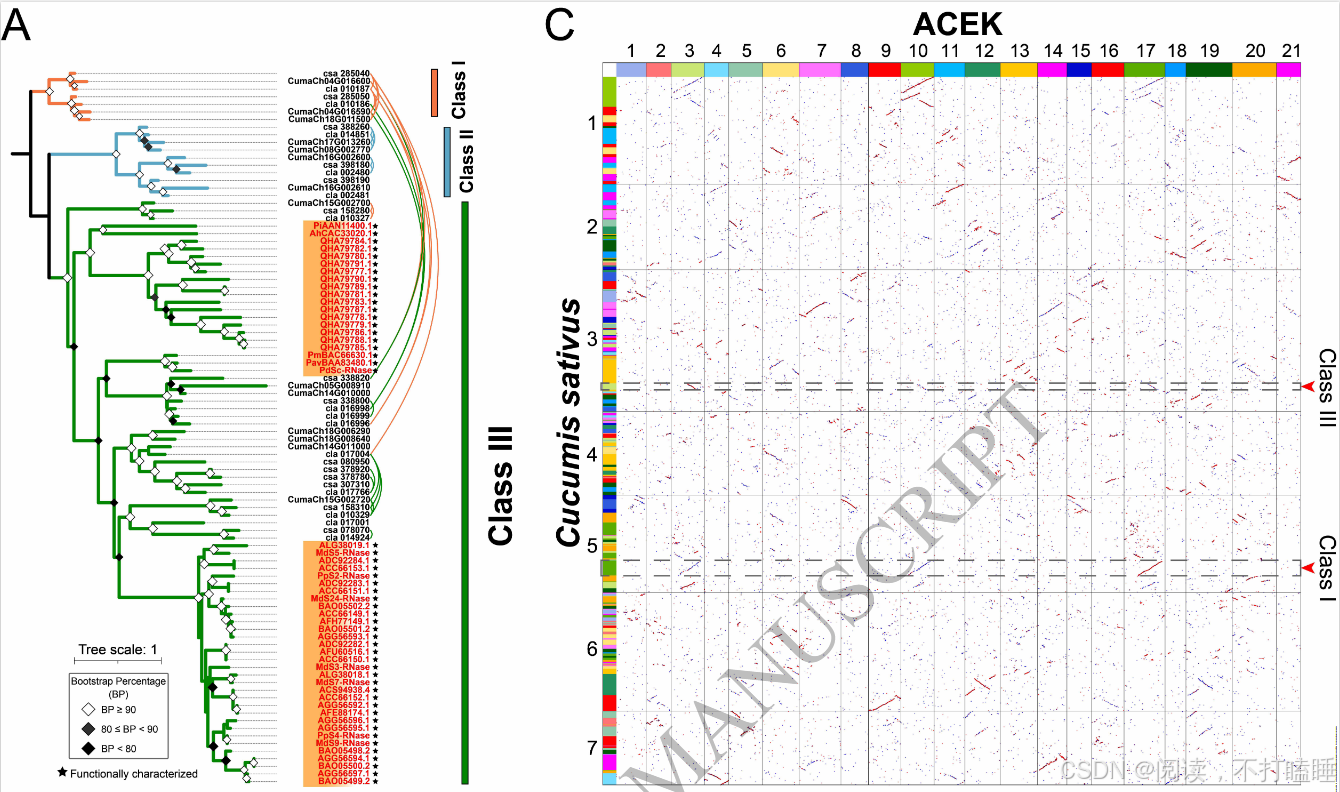
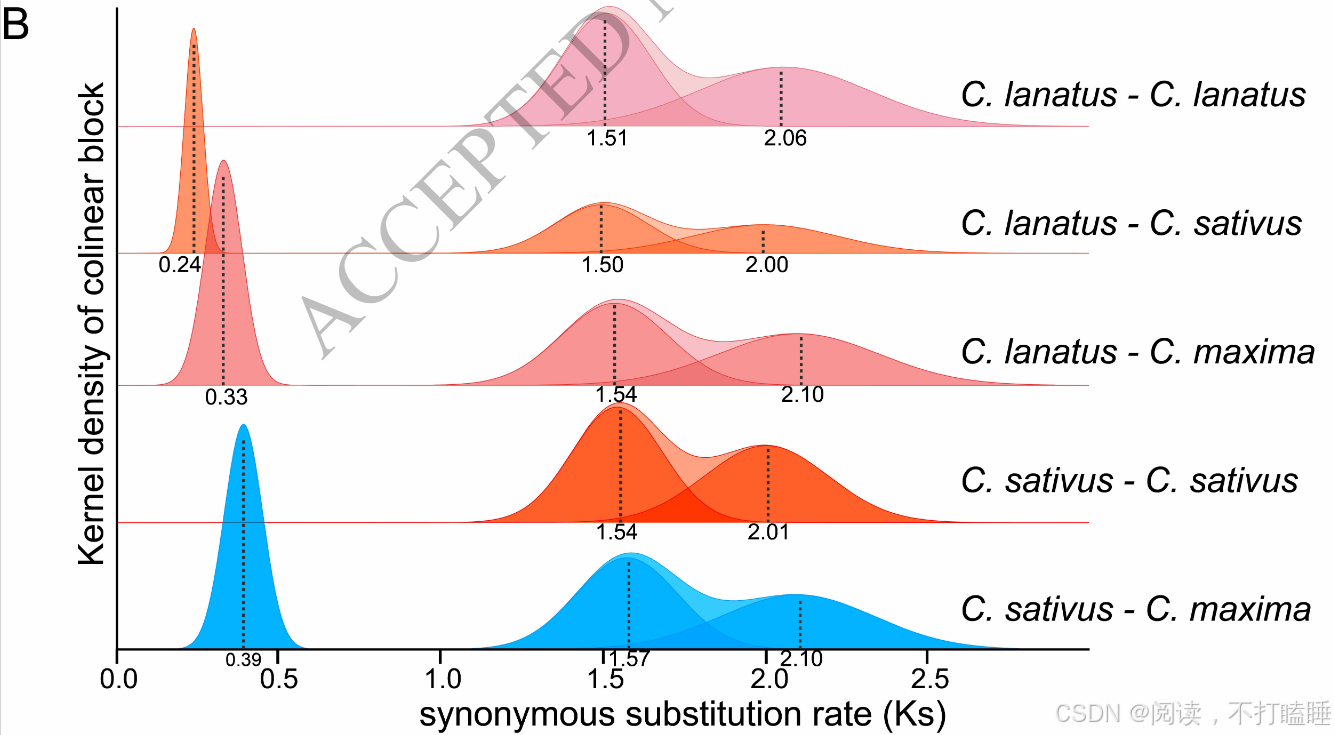
图4
值得注意的是,S-RNase基因和S-like RNase基因在被子植物中发生了谱系特异性的基因转座现象,并伴随着转座元件(TE)活性的显著增强
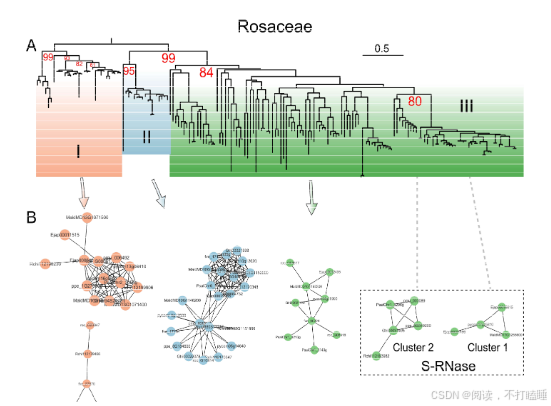
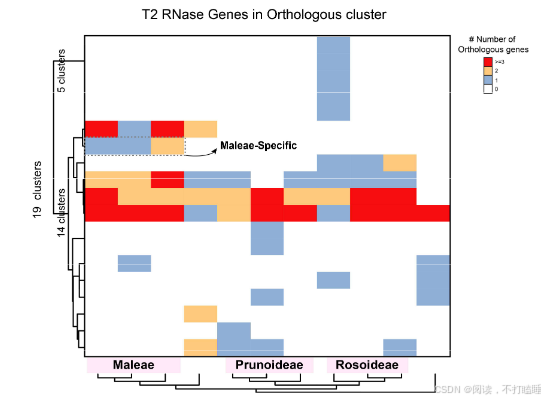
我们从RNase T2基因家族的共线性网络中鉴定出67个共线性群。来自蔷薇科、芸香科、茄科及锦葵科的III类基因(含S-RNase)形成特异于科的独立共线性簇,而Class Ⅱ基因(S-like RNase基因)在十字花科(Brassicaceae)、葫芦科(Cucurbitaceae)和禾本科(Poaceae)中也表现出类似的科特异性共线性簇(图5A,附表S7)。
以11种代表性蔷薇科植物的T2 RNase基因为例,我们进行了详细的共线性网络分析。该共线性网络包含138个节点和239条边,共识别出12个簇(附表S8-S10)。通过系统发育和共线性分析,阐明了它们的进化关系(图5B,附图S17-S18)。值得注意的是,苹果族物种(苹果、西洋梨、枇杷)的S-RNase基因相较于蔷薇科其他类群物种(星草梅、梅亚科和蔷薇亚科物种),聚类在特定的共线性背景和直系同源群组中(图5B,附图S19-S21,附表S11-S12)。除了蔷薇科外,我们还在茄科(图5C)和芸香科(图5D)物种中发现了类似的谱系特异性S-lile RNase基因的转座现象。
这些发现表明,在真双子叶植物谱系中,Class Ⅲ-A类S-RNase基因和S-like RNase基因频繁发生基因转位现象。进一步观察发现,在多个植物基因组中,包括苹果(蔷薇科)、柚子(芸香科)、番茄(茄科)、金鱼草(车前科)和中粒咖啡(茜草科)中,长末端重复序列(LTR)反转录转座子和末端反向重复(TIR)元件与S-RNase基因相邻。这些结果印证了转座元件对S-RNase和S-like RNase基因谱系特异性转位的影响,以及Class Ⅲ基因共线性降低的现象。
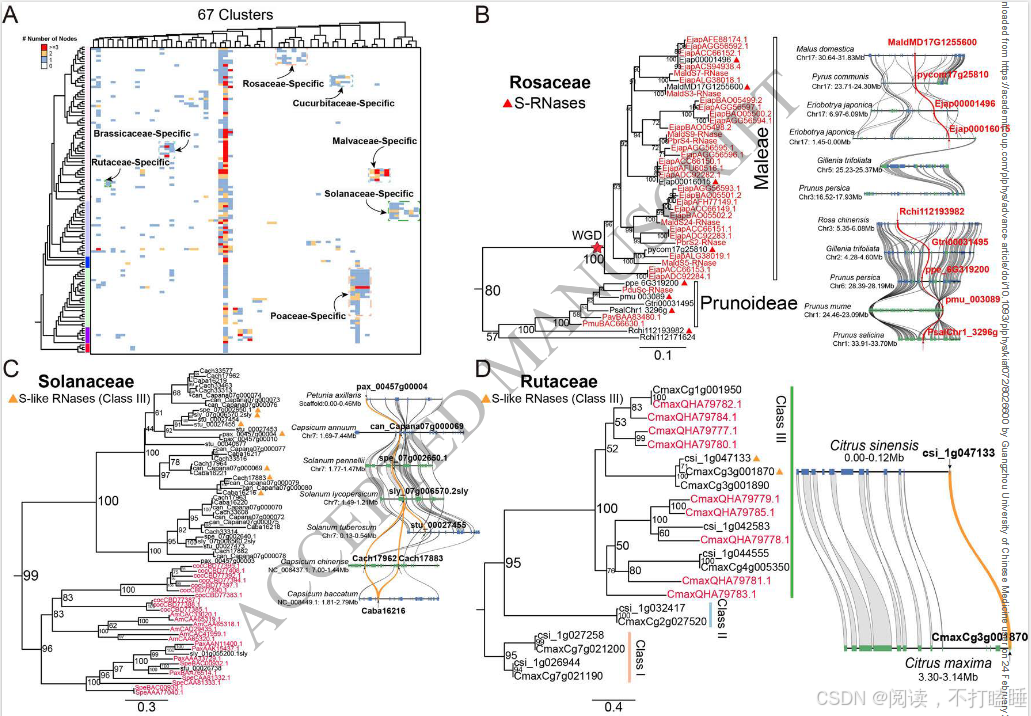
图5
附图S17:蔷薇科RNase T2基因家族的系统发育树。
附图S18:蔷薇科RNase T2基因家族的微共线性网络。(A) 11个蔷薇科基因组RNase T2基因家族的共线性网络。(B) 蔷薇科T2 RNase基因的最大似然系统发育树及其共线性关系。
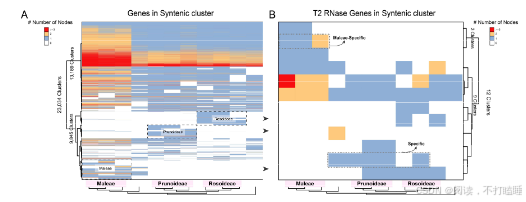
附图S19:11种蔷薇科植物微共簇的系统基因组分析。(A) 11个蔷薇科基因组的所有共线簇的谱图(按每组内的Jaccard距离聚类)。(B) 分析的蔷薇科基因组中RNase T2基因家族的共线性簇图谱。
附图S20:蔷薇科T2 RNase基因的最大似然系统发育树及相应的共线性簇。(A) 11个蔷薇科基因组RNase T2基因家族的最大似然基因树。(B) 蔷薇科T2 RNase基因共线簇的可视化。
附图S21:11种蔷薇科植物同源基因簇的系统基因组分析。
讨论
配子体自交不亲和(GSI)是植物的一种生殖机制,通过花粉识别和排斥来阻止自交受精,促进遗传多样性。S-RNases是该系统的关键组成部分,作为雌蕊表达的基因,它们降解不亲和花粉RNA以维持自交不亲和性。然而,S-RNases的起源一直是个谜。通过揭示它们的基因组历史和和阐明它们在不同物种间的多样化过程来了解它们的进化,可为植物生殖生物学提供关键的见解。通过阐明花粉识别-排斥系统的基因组基础,增强了我们对物种特异性生殖适应和替代交配策略进化的理解,尤其是对那些GSI系统缺失类群(如单性花植物)的植物。
Class Ⅲ- A S-like RNase基因和Class Ⅰ T2 RNase基因之间存在保守的共线性
系统发育共线性分析正成为解析多基因组间保守基因排列、推断功能关系及探究基因进化谱系的关键工具(Ruelens等,2013;Zhao等,2017;Schultz等,2023)。尽管蔷薇科、茄科和芸香科等植物的S-RNase基因已有大量研究,但我们的研究揭示了葫芦科Class Ⅲ-A类S-like RNase与功能表征的S-RNase(图3和S7)和I类T2 RNase(图2)之间的保守共线关系。这些结果显著丰富了我们对复杂进化轨迹的理解,揭示了在葫芦科物种基因组中高度保守的共线性中发现的基因组“化石”(即先前进化阶段的遗传遗迹)这一重要现象。
植物界的大多数物种传统上都表现出两性花(Matthews & Endress, 2004)。然而,各种抑制自花受精机制的进化进一步证实了开花植物促进异花授粉的内在倾向。这促进了遗传多样性的增加和后代的适应性。葫芦科由重要的蔬菜和瓜类组成,其大多数成员物种中表现为单性花,这表明S-RNase基因与自交不亲和的丧失之间存在潜在联系(Boualem etal., 2015)。似乎可以推测,葫芦科物种已经进化出防止自花授粉的机制,例如单性花同株、雄蕊先熟和雌雄异株(Steinbachs & Holsinger, 2002)。因此,在某些植物科中普遍存在的基于S-RNase的配子体自交不亲和性(GSI)机制,可能在葫芦科中不那么重要(Boualem et al., 2015)。
在这一进化背景下,葫芦科物种中基因组背景(共线性)的高度保守性为Class Ⅲ-A基因(包括S-RNase基因)和Class Ⅰ T2 RNase基因起源于真双子叶植物的γ三倍化事件提供了有力证据。研究强调,葫芦科植物单性花的出现以及缺乏基于S-RNase的配子体自交不亲和性(GSI)机制(或类似的生殖系统)可能是这种基因组保守性的重要因素。这些发现为理解基因进化机制和植物生殖系统适应性提供了宝贵的见解。
转座元件对S - RNase进化的影响
分散重复模式对Class Ⅲ基因的贡献最大,这表明它们频繁发生转座。此外,我们在蔷薇科、茄科和芸香科等科中鉴定出Class Ⅲ-A基因的谱系特异性基因簇,暗示S-RNase基因和S-like RNase基因存在显著的谱系依赖性转座事件。蔷薇科苹果族特异性基因簇似乎与该族特异性全基因组复制事件相一致。
转座元件常被认为能够诱导基因分散和转座,从而影响基因组成、功能以及植物基因组的进化(Lisch, 2013)。在S-RNase启动子附近插入一个MITE转座子导致芸香料谱系中SI特征的丧失(Hu et al., 2024)。长末端重复序列转座子与S-RNase基因的邻近性,强调了这些TEs促进基因重排和S-RNase基因转座的潜力,特别是在蔷蔷科谱系中,它们可以通过将基因引入新的基因组环境,或将其从初始位置移除,从而促进苹果族和梅亚科中两个谱系特异性基因组背景的形成。
有趣的是,在配子体自交不亲和性(GSI)系统中,蔷薇科的两个主要分支(苹果族和梅亚科)表现出不同的花粉识别机制,这种多样性似乎是由它们基因组共线性背景的分化所驱动的(Akagi等人,2016;Ashkani & Rees, 2016;Fujii等人,2016;Matsumoto & Tao, 2016)。转座元件插入到S-RNase基因中引入的显著基因组差异,很可能是造成花粉识别过程中机制多样性的原因。
结论
先前的研究表明,Class Ⅰ T2 RNase基因存在于所有主要的陆地植物谱系中,起源于苔藓植物,而Class Ⅱ T2 RNase基因出现较晚,仅限于种子植物(Ramanauskas & igiki, 2017)。在此基础上,我们的研究结合了系统基因组学方法和基于共线性的分析,提出了一个关于S-RNase基因起源和进化轨迹的详细模型(借鉴了上面讨论的观察结果(图6))。我们发现,葫芦科的Class Ⅲ-A类S-like RNase基因和Class Ⅰ T2 RNase基因是核心真双子叶植物γ三倍化事件的重复产物。此外,我们强调了在不同被子植物谱系中形成S-RNases和S-like RNases的不同基因组景观。这些研究结果丰富了我们对T2 RNase基因进化的理解,进一步加深了对植物生殖生物学和进化遗传学的认识。
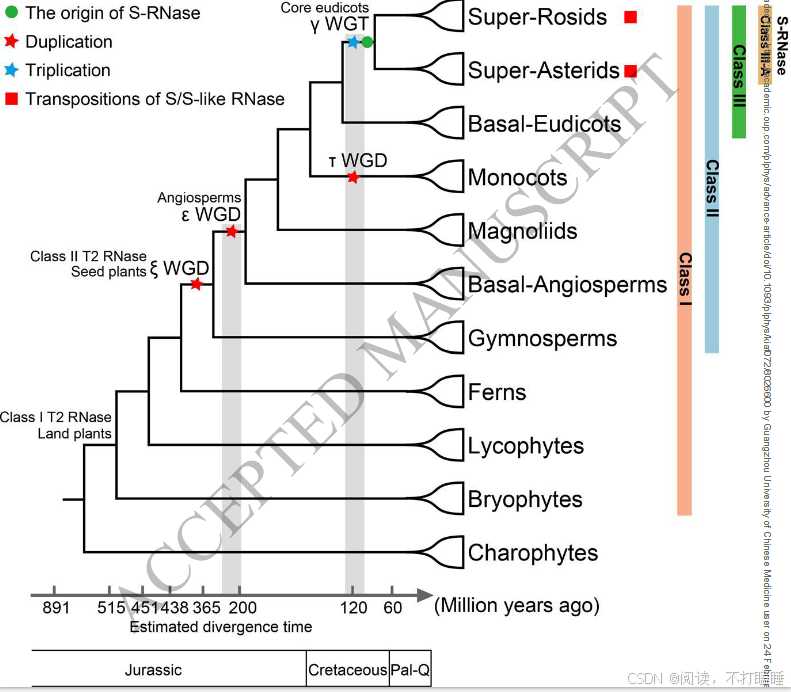
图6
材料与方法
植物基因组资源及RNase T2家族基因的鉴定
该研究中,我们分析了来自不同分类单元的130个完全测序和注释的植物参考基因组,包括66个超蔷薇类植物、23个超菊类植物、5个基部真双子叶植物、25个单子叶植物、6个木兰类植物和4个基部被子植物。在基部被子植物中,即在单子叶植物和真双子叶植物分化之前,我们纳入了以下物种:无油樟、芡实、蓝星睡莲和侏儒睡莲(附图S1)。
基因组注释和编码序列从NCBI、Ensemble、GigaDB、CoGe、Phytozome等已建立的数据库以及GDR、Sol Genomics Network、Citrus Genome Database等特定数据库中下载(详细链接参见附表S1)。我们处理了注释的蛋白质序列和基因组注释文件,以包含每个基因组中的基因位置数据。物种名称被缩写为三个或四个字母的代码,并与各自的蛋白质ID配对。基因组的详细信息及其相关信息见附表S1。
根据以往的研究(Lv et al. 2022; Zhao et al. 2022; Zhu et al. 2020; Zhu et al. 2023),我们利用Pfam数据库中T2 RNase结构域(PF00445)的种子比对文件构建了一个隐马尔可夫模型(HMM)文件,从而鉴定T2 RNase家族基因。我们首先使用HMMER3.0(Finn et al., 2011)以默认的包含阈值(E值 < 1E-3)来初步识别潜在的候选基因。为了确保结果的完整性和准确性,我们进一步利用SMART数据库(http://www.smart.embl-heidelberg.de/)和Pfam(http://pfam.xfam.org/)验证蛋白质序列中是否存在特征性的T2 RNase结构域。此外,我们还使用InterProScan(版本5.60)对蛋白质序列进行分析,参数设置为(-appl Pfam, SMART, SuperFamily -dp -f tsv -goterms -iprlookup)(Jones et al., 2014),并仅保留了含有T2 RNase结构域的序列。相关性分析和统计检验是通过R(版本4.2.3)中的“stats”包完成的。关于130种被子植物基因组中T2 RNase基因家族成员的完整列表,请参阅附表S2。
系统发生重建
我们从GenBank(https://www.ncbi.nlm.nih.gov/genbank/)获取了已报道的S-RNase基因和S-like RNase基因的氨基酸序列,并将其列在附表S3中。对于包含130种被子植物的RNase T2基因家族的大规模系统发育基因树,我们使用MAFFT(版本7.475)对全长氨基酸序列进行比对,参数设置为(-localpair –maxiterate 1000 --thread 20 –reorder)并采用L-INS-I策略(Katoh & Standley, 2013)。随后,我们在MEGA(版本11)中手动校对比对结果,去除空缺位点并丢弃缺乏保守基序的序列(Tamura et al., 2011)。使用IQ-TREE中的ModelFinder算法(-m MF -T AUTO)确定最佳氨基酸替代模型。WAG+R8模型被确定为适用于130种物种中RNase T2基因家族的最佳模型(Kalyaanamoorthy et al., 2017; Nguyen et al., 2015)。最大似然(ML)系统发育树是通过IQ-TREE(版本2.0.3)计算得出的,参数设置为:-m WAG+R8 -alrt 1000 -bb 1000 -nt 10(Lin et al., 2013)。对于11种蔷薇科物种的小规模系统发育树,分析使用IQ-TREE(版本2.0.3)进行,参数设置为:-m VT+R6 -alrt 1000 -bb 1000 -nt 10。最终的最大似然基因树通过在线工具iTOL v5(https://itol.embl.de/)和FigTree v1.4.4(http://tree.bio.ed.ac.uk/software/figtree/)进行可视化和注释(Letunic & Bork, 2021)。
基因复制模式的确定
每个物种中T2 RNase基因的起源被划分为五种不同的基因复制模式:全基因组复制(WGD)、串联复制(TD)、邻近复制(PD)、转座复制(TRD)和分散复制(DSD)。这种分类是通过使用DupGen_finder(https://github.com/qiao-xin/DupGen_finder)完成的,采用的是默认参数(Qiao et al., 2019; Wang et al., 2012)。
具体而言,串联复制(TD)指的是位于同一染色体上且彼此相邻的同源基因拷贝。邻近基因对(PD)是指在同一染色体上由10个或更少基因分隔的非串联基因对。转座复制(TRD)通常被标记为“转座”,涉及远端位置的基因对,其中一个基因是共线性的,而另一个基因是非共线性的。这种排列形成了一个由祖先位点和新位点组成的基因对。分散复制(DSD)事件则产生两个既不相邻也不共线性的基因拷贝。
那些不属于全基因组复制(WGD)、串联复制(TD)、邻近复制(PD)或转座复制(TRD)的剩余重复基因对被视为分散重复(DSD)。重复基因的优先级被设定为:全基因组复制(WGD)> 串联复制(TD)> 邻近复制(PD)> 转座复制(TRD)> 分散重复(DSD)。单拷贝基因由于在目标物种中缺乏同源对应物,因此不被归类为重复类型。这种分类的结果提供在附表S4-S5中。
共线性网络分析和共线性簇的系统发育基因组学分析
Zhao和Schranz(2017)开发的SynNet构建流程(https://github.com/zhaotao1987/SynNet462 Pipeline)被用于构建共线性网络,该共线性网络涵盖了所研究被子植物基因组内和基因组间的共线性比较,以及所有基因之间的共线性关系。第一步是使用Diamond v3.3.2(Buchfink等人,2015)对所研究的130种被子植物物种的蛋白质序列进行全对全的相互比较。接下来,我们使用MCScanX在默认参数下(共线性区块的最小匹配大小为5个基因,允许的最大间隔为25个基因)计算所有成对基因组组合之间的基因组共线性(即多个物种基因组中保守的基因顺序和内容)(Wang等人,2012)。
随后,利用所有候选T2 RNase基因的列表,从整个共线性网络数据库中提取了一个专门针对T2 RNase基因的子网络(附表S6)。在这个子网络中,每个节点代表一个基因,边表示基因之间的共线性连接,而边的长度没有意义(无权重)。为了对网络进行聚类,我们采用了Infomap算法(基于图划分框架)(Rosvall & Bergstrom, 2008)。R包“igraph”被用于执行网络统计分析。最终的T2 RNase基因家族共线性网络通过Cytoscape v.3.8.0(Shannon等人,2003)和Gephi(Bastian等人,2009)进行可视化和分析。
我们使用iTOL v5在线平台(https://itol.embl.de/)将共线性关系映射到构建的系统发育树上。我们评估了每个共线性群组内不同物种的基因拷贝数。这些分析结果展示了每个基因组中每个共线性簇的T2 RNase基因数量,并通过“Jaccard”距离和“ward.D”聚类方法进行整理和可视化。共线性网络分析的详细信息和结果分析提供在附表S7-S10中。
我们使用JCVI软件包中实现的MCscan(Python版本)(https://github.com/tanghaibao/jcvi/wiki/MCscan-(Python-version)展示了不同物种间显著的微共线性背景(Tang等人,2015)。更多细节可以在附表S11中找到。
直系同源簇的直系同源鉴定和系统发育基因组学分析
本研究中的蔷薇科数据集包括11个基因组,涵盖了三个亚科:梅亚科(苹果、梨、枇杷、星草梅、桃、梅和李)、蔷薇亚科(黑莓、月季和草莓以及仙女木亚科(亮黄仙女木)。我们使用OrthoFinder(版本2.5.2),并设置参数(-S diamond -M msa -A mafft -I 1.5),从这11种蔷薇科植物的蛋白质序列中鉴定同源基因群组(orthogroups)(Emms & Kelly, 2019)。
通过系统发育组学分析,本研究获得包含28,069个非冗余多基因簇(直系同源群)的特征矩阵。每个同源群组和每个共线性簇都被注释了所代表的物种数量,以确定所有簇之间的差异性。随后,我们计算了一个差异性指数,并使用“ward.D”方法进行了层次聚类。最终的聚类热图通过“pheatmap”进行可视化,直系同源群组的详细信息可以在附表S12中找到。
同义替换率( Ks )计算,转座子鉴定
对于葫芦科基因组,我们使用WGDI v.0.62工具包(Sun et al., 2022)生成了Ks点图。Ks值的密度分布曲线通过Kspeaks模块(-kp)创建,并使用PeaksFit模块(-pf)进行多峰值拟合。从这些曲线中估计的平均峰值用于推断全基因组复制(WGD)事件的时间。对于ACEK(Ancestor-Collinear-Expansion-Karyotype)绘图,使用“icl”参数识别ACEK与特定物种之间的共线性基因,并使用“-km”参数进行ACEK绘图。最后,使用WGDI的“-d”参数和祖先基因组(Cucumis sativus)生成同源点图。
我们使用了广泛的从头转座子注释工具(EDTA v1.9.4),并设置了参数:--sensitive 1 --anno 1,以鉴定转座元件。在本研究中,我们仅考虑了完整的转座元件。关于葫芦科基因组中比较的Ks值以及鉴定到的转座元件的详细信息可以在附表S13中找到。
基因表达分析
本研究的RNA-Seq数据来自NCBI SRA数据库(<https://www.ncbi.nlm.nih.gov/sra>)。我们使用Fastp v.0.12.4对原始数据进行质量控制,并设置参数(-f 12 -F 12 -l 50),以确保数据的高质量(Chen等人,2018)。随后,我们使用了Kallisto v.0.46.2将样本中过滤后的高质量读段比对到参考基因组中注释基因的编码序列(CDS)(Bray等人,2016)。我们使用TPM值来量化组织中基因的表达水平。关于所用RNA-Seq数据的详细信息可在附表S14中找到。
不同物种中S -位点的注释
S-位点的注释是基于以往研究(Lv et al. 2022;Zhao et al. 2022)。GSI S-位点包括S-RNase基因和紧密连锁的SLFs。关于鉴定到的S-位点的详细信息见附表S15。


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









