






安装和加载包
将 转变为
转变为
创建工作目录,复制这两个文件进去。
#加载包和注释文件
library(tidyverse)#加载包
load("gene_annotation_2022.rda")#导入gene注释文件
#加载TCGA下载的rda文件
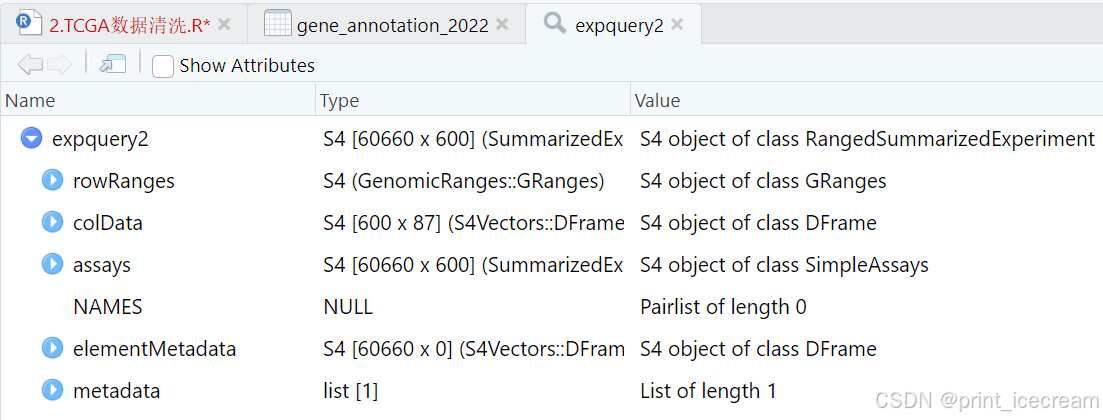
load("luad.gdc_2022.rda")#导入文件,rda格式文件也可直接从文件夹双击导入luad.gdc_2022.rda文件得到expquery2数据
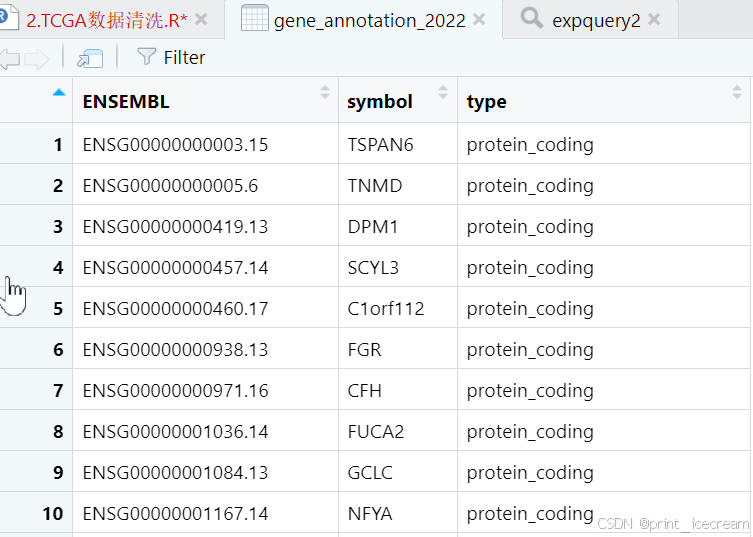
提出TCGA数据
#提出TCGA原始数据、给与列名、给与行名
counts <- expquery2@assays@data@listData[["unstranded"]]#行名123456 列名V1V2V3
colnames(counts) <- expquery2@colData@rownames #列名= 样本名
rownames(counts) <- expquery2@rowRanges@ranges@NAMES #行名= 探针名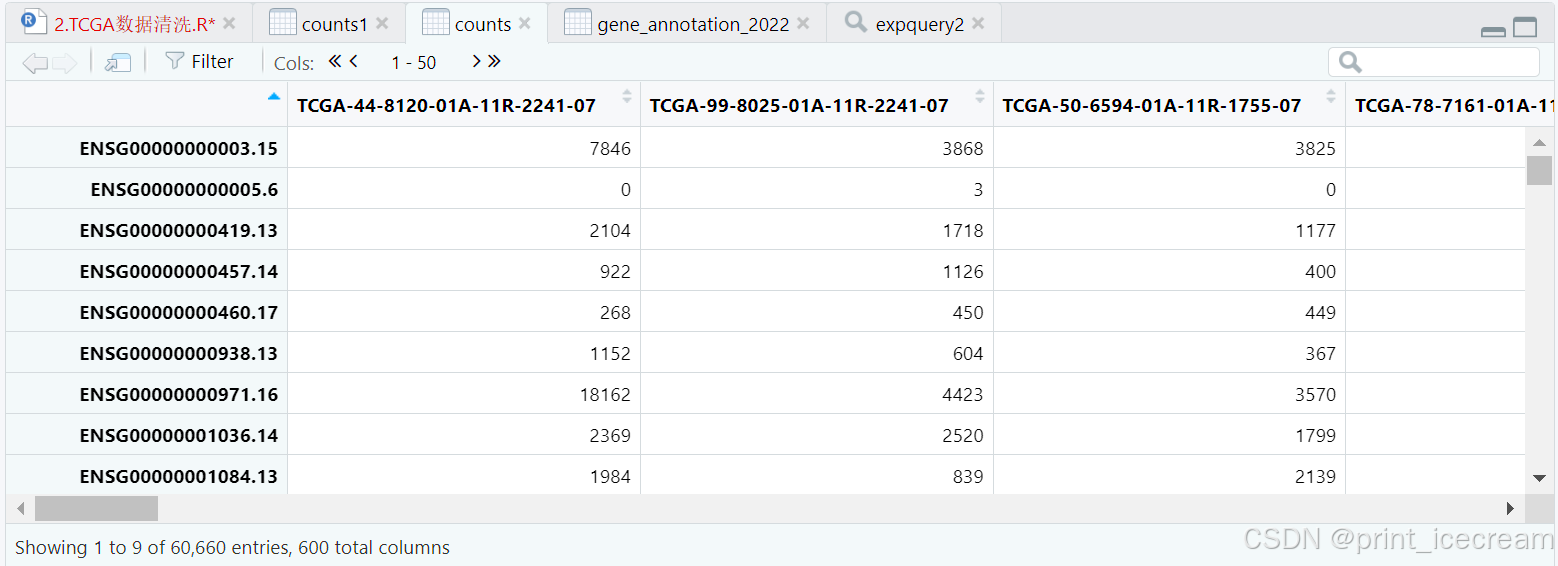
转为数据框格式
#下面代码一样
counts1 <- counts
counts1 <- as.data.frame(counts1) #数据框
counts1 <- rownames_to_column(counts1,var = 'ENSEMBL') #数据框的行名转换为数据框的一列
counts1 <- inner_join(counts1,gene_annotation_2022,"ENSEMBL") #合并counts1与gene_annotation_2022
#按照counts1的ENSEMBL去排序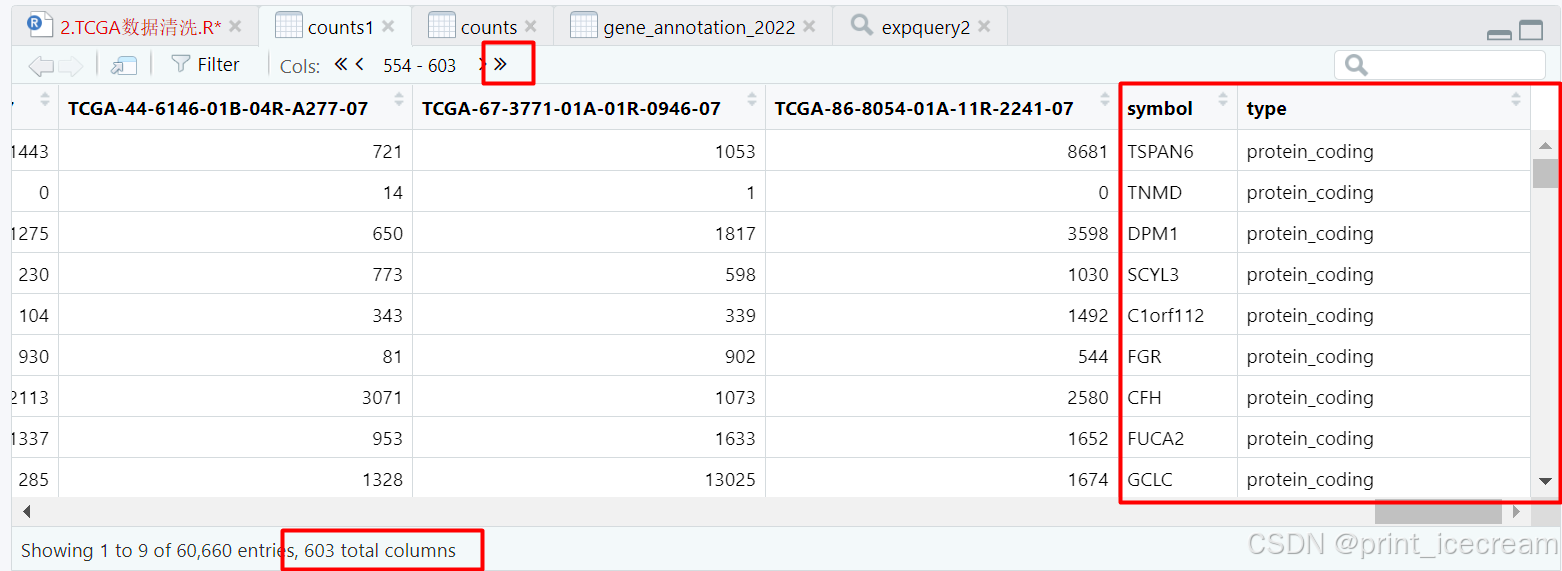
完成行名的整理
#查重:去除重复symbol
counts1 <- counts1[!duplicated(counts1$symbol),]#美元符号
#symbol列变行名
rownames(counts1) <- NULL #无
counts1 <- column_to_rownames(counts1,var = 'symbol')
#只保留编码基因
table(counts1$type)# 注19934
counts <- counts1[counts1$type == "protein_coding",]
#删除第一列与最后一列
counts <- counts[,-c(1,ncol(counts))]
整理列名:按01A和11A排布
# 把TCGA barcode切割为16位字符,并去除重复样本
# substr(字符串, 起始位置, 结束位置)
colnames(counts) <- substring(colnames(counts),1,16)
#去重
counts <- counts[,!duplicated(colnames(counts))]
#查看多少01A
table(substring(colnames(counts),14,16))
# 保留01A (注:可通过table(substring(colnames(counts),14,16))查看样本类型)
# 14到16是01A肿瘤
counts01A <- counts[,substring(colnames(counts),14,16) == c("01A")]
# 保留11A
counts11A <- counts[,substring(colnames(counts),14,16) == c("11A")]
table(substring(colnames(counts01A),14,16))
table(substring(colnames(counts11A),14,16))
#cbind之前需要确认两个数据框的行名
identical(rownames(counts01A),rownames(counts11A))
counts <- cbind(counts01A,counts11A) #重新革新counts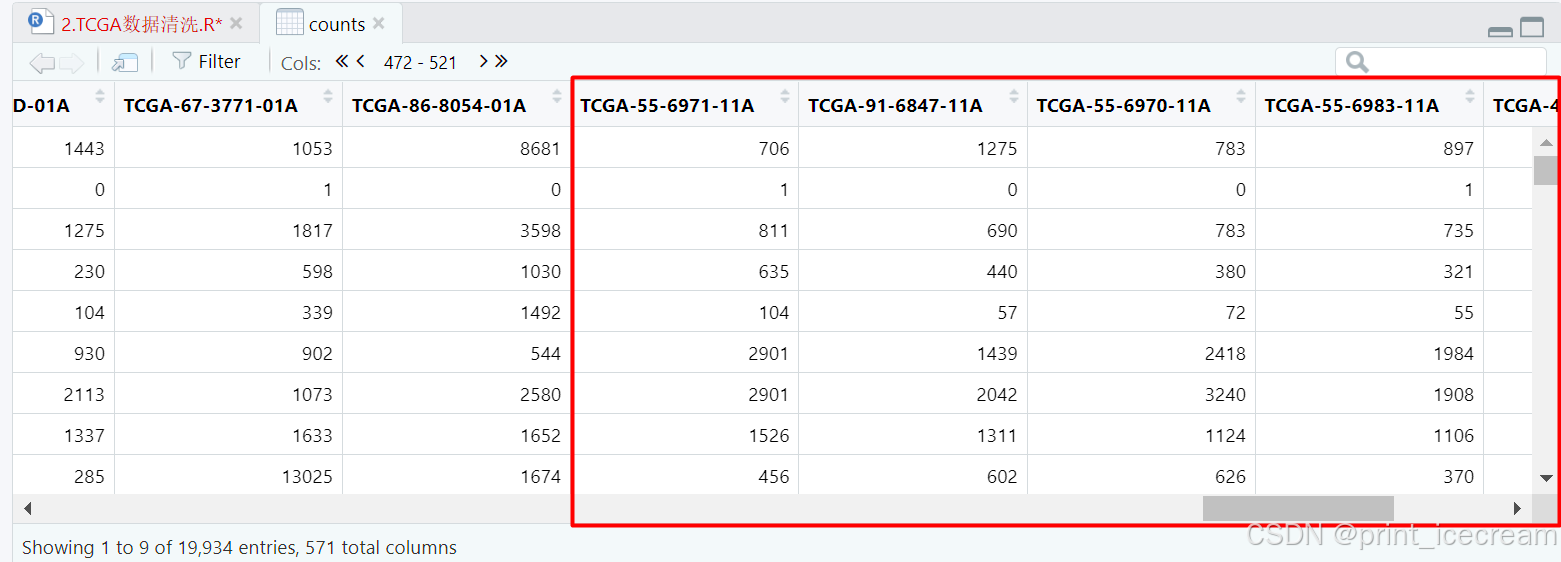
保存全部、01A、11A的txt
#保存数据:01A肿瘤 11A正常
write.table(counts,"counts.txt",sep = "\t",row.names = T,col.names = NA,quote = F)
write.table(counts01A,"counts01A.txt",sep = "\t",row.names = T,col.names = NA,quote = F)
write.table(counts11A,"counts11A.txt",sep = "\t",row.names = T,col.names = NA,quote = F)
完整代码
#加载包和注释文件
library(tidyverse)#加载包
library(tibble)
load("gene_annotation_2022.rda")#导入gene注释文件
#加载TCGA下载的rda文件
load("luad.gdc_2022.rda")#导入文件,rda格式文件也可直接从文件夹双击导入
#提出TCGA原始数据、给与列名、给与行名
counts <- expquery2@assays@data@listData[["unstranded"]]#行名123456 列名V1V2V3
colnames(counts) <- expquery2@colData@rownames #列名= 样本名
rownames(counts) <- expquery2@rowRanges@ranges@NAMES #行名= 探针名
#下面代码一样
counts1 <- counts
counts1 <- as.data.frame(counts1) #数据框
counts1 <- rownames_to_column(counts1,var = 'ENSEMBL') #数据框的行名转换为数据框的一列
counts1 <- inner_join(counts1,gene_annotation_2022,"ENSEMBL") #合并counts1与gene_annotation_2022
#按照counts1的ENSEMBL去排序
#查重:去除重复symbol
counts1 <- counts1[!duplicated(counts1$symbol),]#美元符号
#symbol列变行名
rownames(counts1) <- NULL #无
counts1 <- column_to_rownames(counts1,var = 'symbol')
#只保留编码基因
table(counts1$type)# 注19934
counts <- counts1[counts1$type == "protein_coding",]
#删除第一列与最后一列
counts <- counts[,-c(1,ncol(counts))]
# 把TCGA barcode切割为16位字符,并去除重复样本
# substr(字符串, 起始位置, 结束位置)
colnames(counts) <- substring(colnames(counts),1,16)
#去重
counts <- counts[,!duplicated(colnames(counts))]
#查看多少01A
table(substring(colnames(counts),14,16))
# 保留01A (注:可通过table(substring(colnames(counts),14,16))查看样本类型)
# 14到16是01A肿瘤
counts01A <- counts[,substring(colnames(counts),14,16) == c("01A")]
# 保留11A
counts11A <- counts[,substring(colnames(counts),14,16) == c("11A")]
table(substring(colnames(counts01A),14,16))
table(substring(colnames(counts11A),14,16))
#cbind之前需要确认两个数据框的行名
identical(rownames(counts01A),rownames(counts11A))
counts <- cbind(counts01A,counts11A) #重新革新counts
#保存数据:01A肿瘤 11A正常
write.table(counts,"counts.txt",sep = "\t",row.names = T,col.names = NA,quote = F)
write.table(counts01A,"counts01A.txt",sep = "\t",row.names = T,col.names = NA,quote = F)
write.table(counts11A,"counts11A.txt",sep = "\t",row.names = T,col.names = NA,quote = F)

















 671
671

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









