






 研究通过定量微生物组方法分析了肉鸡肠道细菌和真菌的发育过程,发现肠道微生物在7日龄达到平台期,呈现肠段特异的群落类型。相对丰度分析与绝对丰度分析在揭示微生物变化和相互关系时存在差异。微生物定植能力不同,形成不同的发育模式。此外,肠道微生物与血清代谢物有密切关联,尤其前肠微生物和真菌对代谢物变化影响显著。
研究通过定量微生物组方法分析了肉鸡肠道细菌和真菌的发育过程,发现肠道微生物在7日龄达到平台期,呈现肠段特异的群落类型。相对丰度分析与绝对丰度分析在揭示微生物变化和相互关系时存在差异。微生物定植能力不同,形成不同的发育模式。此外,肠道微生物与血清代谢物有密切关联,尤其前肠微生物和真菌对代谢物变化影响显著。
点击蓝字 关注我们
定量微生物组分析揭示鸡肠道微生物发育轨迹及其与宿主代谢的关系

iMeta主页:http://www.imeta.science
研究论文
● 原文链接DOI: https://doi.org/10.1002/imt2.105
● 2023年4月25日,中国农业大学胡永飞和呙于明团队在 iMeta 在线发表了题为“Quantitative microbiome profiling reveals the developmental trajectory of the chicken gut microbiota and its connection to host metabolism ” 的文章。
● 本研究以绝对定量的方式描绘鸡肠道微生物群发育过程,肠道微生物与血清代谢物之间的关联分析进一步凸显了肠道微生物对鸡生长发育的影响。
● 第一作者:封雨晴
● 通讯作者:胡永飞 (huyongfei@cau.edu.cn)、呙于明(guoyum@cau.edu.cn)
● 合作作者:张美红、刘艳、杨新月、魏福晓、金晓露、刘丹
● 主要单位:中国农业大学 动物科学技术学院 动物营养学国家重点实验室
亮 点
● 通过定量微生物组分析揭示肉鸡肠道微生物发育轨迹
● 鸡肠道微生物呈现出肠段特异的群落类型和生长阶段特异的微生物
● 不同鸡肠道微生物定植能力不同,形成不同的发育模式
● 鸡肠道微生物的发育与血清代谢物变化有关
摘 要
揭示鸡肠道微生物的建立和演替过程对于理解其在宿主生理和代谢中的作用至关重要。然而,很少有研究使用定量微生物组分析方法研究鸡肠道微生物绝对丰度的动态变化。我们通过结合高通量测序和定量微生物组的方法,揭示肉鸡肠道中细菌菌群和真菌菌群发育轨迹。研究结果表明,1日龄后鸡肠道微生物形成肠段特异的群落结构,且肠道微生物丰度和多样性在7日龄达到平台期。细菌菌群受“确定性过程”影响较大,而真菌菌群受“随机过程”影响较大。我们在不同肠段都发现肠段特异的微生物,并定义三种微生物发育模式,包括“定植”、“消失”和“核心”。微生物共现网络在不同肠段之间有很大区别,且含有的正相关关系数量多于负相关关系数量。此外,我们进一步分析鸡肠道微生物绝对丰度与宿主血清代谢物之间的关联。非靶向代谢组学时序分析揭示六个变化模式不同的簇。前肠微生物与鸡血清代谢物之间关系更加密切;肠道微生物与脂质和氨基酸代谢密切相关。本研究以绝对定量的方式描绘鸡肠道微生物群发育过程,肠道微生物与血清代谢物之间的关联分析进一步凸显了肠道微生物对鸡生长发育的影响。
视频解读
Bilibili:https://www.bilibili.com/video/BV1Qk4y1L7rx/
Youtube:https://youtu.be/x0Z6Urnw3zo
中文翻译、PPT、中/英文视频解读等扩展资料下载
请访问期刊官网:http://www.imeta.science/
全文解读
引 言
近几十年来,人们对家禽产品的需求量成倍增长,鸡肉已成为全世界消费最多的肉类。鸡肠道微生物影响着宿主免疫、营养物质消化和饲料转化率,这表明肠道微生物对鸡的健康和生产性能是十分重要的。与其他动物类似,鸡肠道微生物主要由细菌、古菌、真菌、病毒等组成。许多研究使用培养或不依赖培养方法研究鸡肠道微生物组成和功能。然而,大多数研究主要精力集中在鸡肠道内细菌,对真菌关注较少,然而真菌在鸡的消化和营养供应等方面也有贡献。此外,与前肠相比,更多研究关注于鸡后肠微生物。
近年来,宿主肠道内微生物建立和演替过程引起广泛关注。肠道微生物在生命早期的发育对宿主健康有着深远影响,包括刺激免疫系统发育和免疫细胞成熟,保持内分泌系统稳态等。鸡肠道内细菌菌群变化与年龄密切相关;在早期阶段,变形菌门在盲肠微生物中占主导地位,随后厚壁菌门占据主导地位。在整个肉鸡生产周期,乳酸菌属是回肠中丰度最高的细菌。鸡肠道内真菌菌群发育的相关研究较少,直到最近一项工作发现前肠真菌菌群的多样性比后肠高,且直到42日龄都没有发现明显的演替趋势。重要的是,这项研究系统比较了真菌和细菌的绝对丰度,并发现真菌在鸡肠道微生物中占比较小。
相对丰度是用来描述群落内微生物在不同时空下的比例,定量微生物组分析则关注微生物绝对丰度的变化。因此,定量微生物组分析使我们能够了解真实的群落变化,有利于鉴别塑造微生物群落结构的生物和非生物因素及肠道微生物和宿主变化之间的关联。此外,微生物数量本身也是一个生态系统标志物,如健康和疾病状态。人类微生物组相关研究已经使用定量微生物组分析来确定与炎症性肠病患者病理生理表现和疾病诊断相关的微生物标志物。基于绝对定量微生物关联分析显示,早产儿的微生物组成动态变化可能是由特定微生物之间相互作用所驱动。目前已开发不同微生物组定量方法,如测定微生物DNA含量、实时荧光定量PCR、流式细胞术和微生物插入技术。与微生物组相对定量方法相比,定量微生物组分析将测序数据转化为微生物绝对丰度矩阵,并在下游分析前确定微生物绝对丰度。
以前研究往往基于16S扩增子测序得到相对丰度结果研究鸡肠道微生物发育情况。然而,人们对绝对丰度下细菌和真菌在鸡肠道发育轨迹并不完全了解。本研究使用定量微生物组方法研究了鸡十二指肠、空肠、回肠和盲肠在42日龄内微生物群落演替情况。我们还使用非靶向代谢组学来研究鸡血清中代谢物随时间变化情况,并构建肠道微生物发育和代谢物间的关联。
结 果
鸡肠道微生物丰度和多样性在7日龄达到平台期
为了研究肉鸡肠道微生物在整个生产周期(1-42日龄)发育轨迹,我们收集总共80只鸡的320个肠道内容物样本,用定量微生物组方法研究四个肠段内细菌和真菌群落演替情况。由于16S rRNA基因V3-V4高变区引物可以同时检测到细菌和古菌,我们也对古菌的动态变化进行分析。研究发现,样品中古菌检出率较低(小于15.0%),在回肠中并没有发现古菌存在(补充材料图S1A)。此外,在检出古菌的样本中,细菌和真菌数量分别是古菌数量的4和1-3个数量级(补充材料图S1B)。这些结果表明,古菌在鸡的肠道中是比较少见的,这与我们以前的发现一致。因此,我们后续重点分析细菌菌群和真菌菌群。
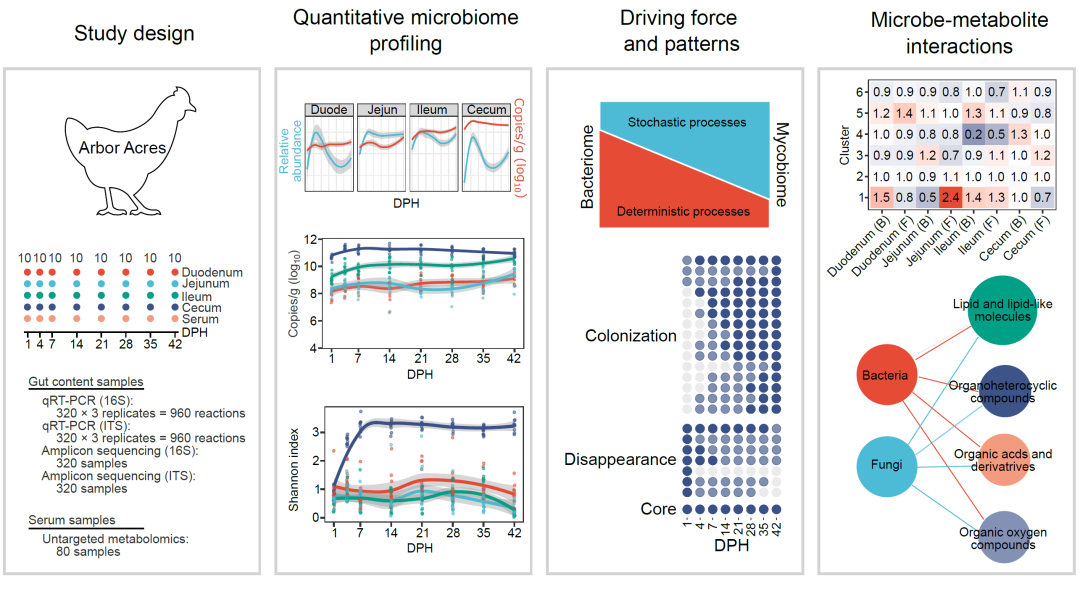
我们发现,细菌和真菌的绝对丰度在7日龄到达平台期,而且细菌数量沿着肠道呈增加的趋势。回肠中真菌数量高于盲肠中真菌数量,特别是在7日龄后(图1A)。细菌绝对丰度在前三个肠段随时间递增,但真菌在四个肠段都呈现出先增后减的趋势。物种组成分析表明,在鸡肠道微生物中总共有25个细菌门和9个真菌门。在门水平上,厚壁菌门和子囊菌门分别是细菌和真菌中主要的门(补充材料图S2)。盲肠中拟杆菌门数量从14日龄开始增加,而真菌担子菌门在大多数肠段中伴随着日龄呈现减少的趋势。值得注意的是,新孵化肉鸡在四个肠段都有很大比例的变形菌门。在属水平上,乳酸菌属是前三个肠段中最主要的属,但在4日龄及以后的盲肠中则不是主要的属(图1B)。链球菌属和埃希氏菌属-志贺菌属在1日龄时占据整个肠道。真菌菌群在组成上则更为复杂。在整个42日龄期间,念珠菌属、镰刀菌属和毛孢子菌属在所有的肠段中占据主导地位,念珠菌属似乎更适应空肠和回肠环境(图1C)。盲肠中细菌菌群多样性比其他三个肠段高,这可能是由于乳酸杆菌在前肠中占主导地位所致。真菌群落多样性随时空变化相对较小(图1D)。然而,盲肠中细菌菌群和真菌菌群多样性在最初7天内急剧增加,这表明此时肠道微生物处于快速定植阶段。肠道微生物组成和多样性变化进一步反映在不同肠段菌群结构差异上(补充材料表S4-S5和图S3,FDR < 0.05)。相邻时间点的样本之间的成对Bray-Curtis距离分析表明,细菌菌群和真菌菌群结构变化更多发生在7日龄前(图1E)。这些结果表明在肉鸡肠道不同肠段有不同微生物组成,在肠道微生物发育过程中,微生物丰度和多样性在7日龄达到一个相对稳定的状态。
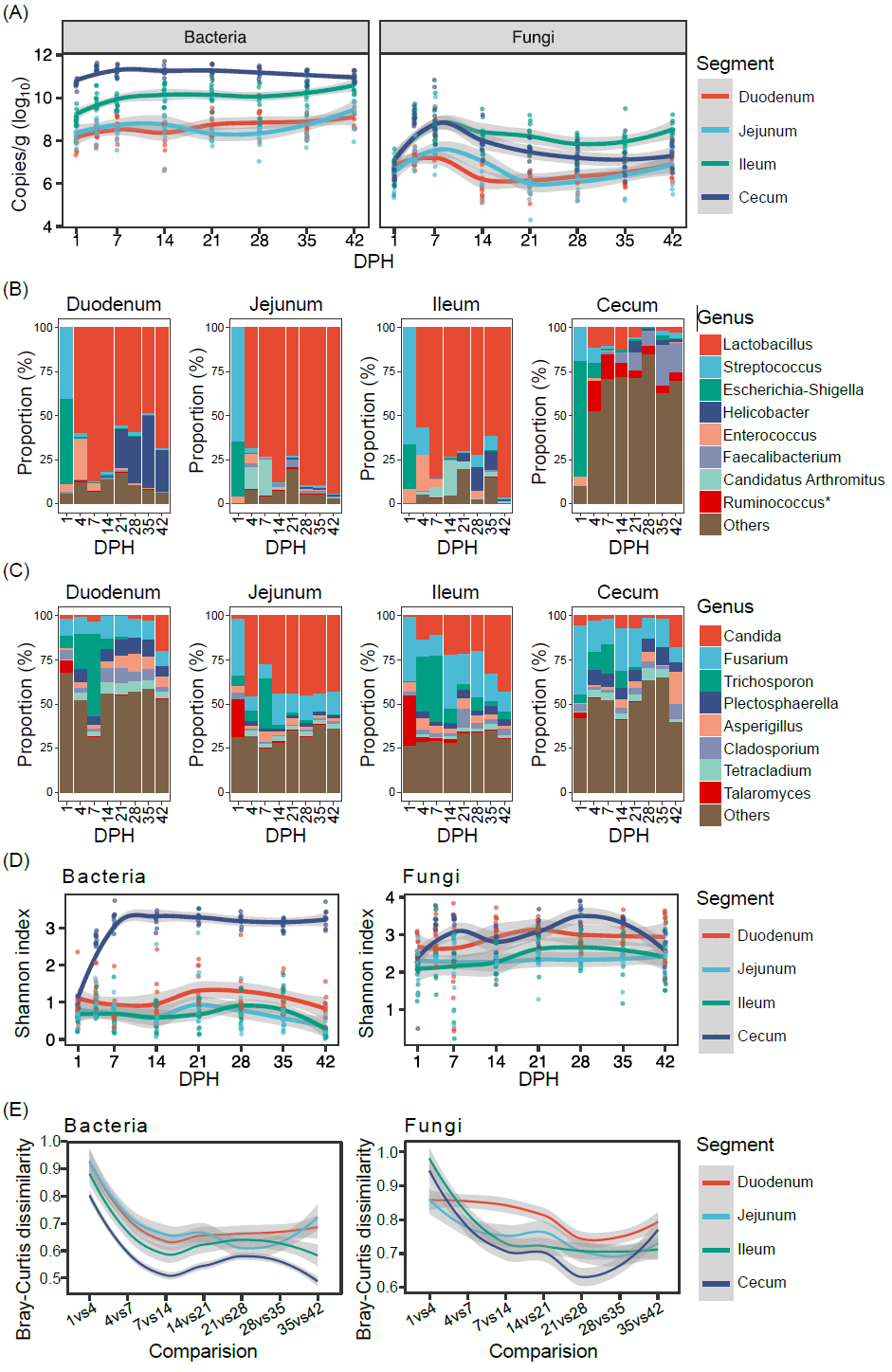
图 1. 鸡肠道微生物绝对丰度和多样性的变化
(A)肉鸡肠道内细菌和真菌在四个肠段中绝对丰度动态变化;肉鸡肠道内细菌(B)和真菌(C)属水平上结构组成,*代表torques group;(D)样本在42日龄内香农指数;(E)相邻时间点样本Bray-Curtis距离比较。DPH: 孵化后天数。
相对丰度揭示的鸡肠道微生物发育和相互关系有偏差
由于微生物丰度在宿主肠道内随时空变化,相对丰度干扰了肠道微生物与宿主特征关系的建立。为确定定量微生物组和相对定量微生物组方法在结果上的差异,我们首先分析主要的门和属的发育轨迹。在这两个分类层级上都呈现出明显的发育轨迹差异。例如,细菌中厚壁菌门在四个肠段中绝对丰度相对稳定。然而,厚壁菌门相对丰度呈现出较大变异,特别是在十二指肠和盲肠中(图2A)。在两种不同方法下,真菌中子囊菌门在四个肠段呈现出相反的发育轨迹。虽然在两种方法下,乳酸菌属的丰度有类似的变化趋势,但定量微生物组分析结果表明7日龄后乳酸菌属绝对数量在不同的肠段中是接近的,但相对定量微生物组结果显示其在盲肠中丰度极低。真菌中念珠菌属也有类似的结果。普氏分析表明,细菌菌群和真菌菌群的绝对丰度和相对丰度是密切相关的(p < 0.001)。然而,细菌菌群和真菌菌群的相关系数分别只有0.622和0.459(图2B)。与绝对丰度相比,相对丰度夸大了个体、肠段和日龄等因素的解释度(图2C)。
基于微生物丰度相关性构建的共现网络被广泛用于肠道微生物分析当中,用于推断和解释微生物间相互关系。为了研究不同定量方法对揭示微生物相互关系的影响,我们分析了不同肠段属水平的Spearman相关性。在定量微生物组和相对定量微生物组网络中都发现了大量明显共同变化的属(补充材料图S4)。四个肠段(十二指肠、空肠、回肠和盲肠)内细菌菌群相关性(正相关+负相关+不相关)在两种方法下结果的一致性分别为82.1%、66.0%、78.7%和86.8%,而在真菌菌群中结果的一致性分别为81.1%、73.8%、78.6%和56.0%(图2D,E)。以定量微生物组结果作为参照,相对定量微生物组方法容易高估细菌菌群间的正相关关系数量(十二指肠402对196,空肠1164对678,回肠1050对725,盲肠668对683),而低估真菌菌群间正相关关系数量(十二指肠400对775,空肠997对1338,回肠865对1044,盲肠1048对2270)。
总的来说,相对定量微生物组方法忽略了微生物绝对数量的增减,不能发现鸡肠道中真正的微生物变化情况。因此,相对定量微生物组方法揭示肠道微生物发育轨迹和其中的微生物相互关系有偏差。
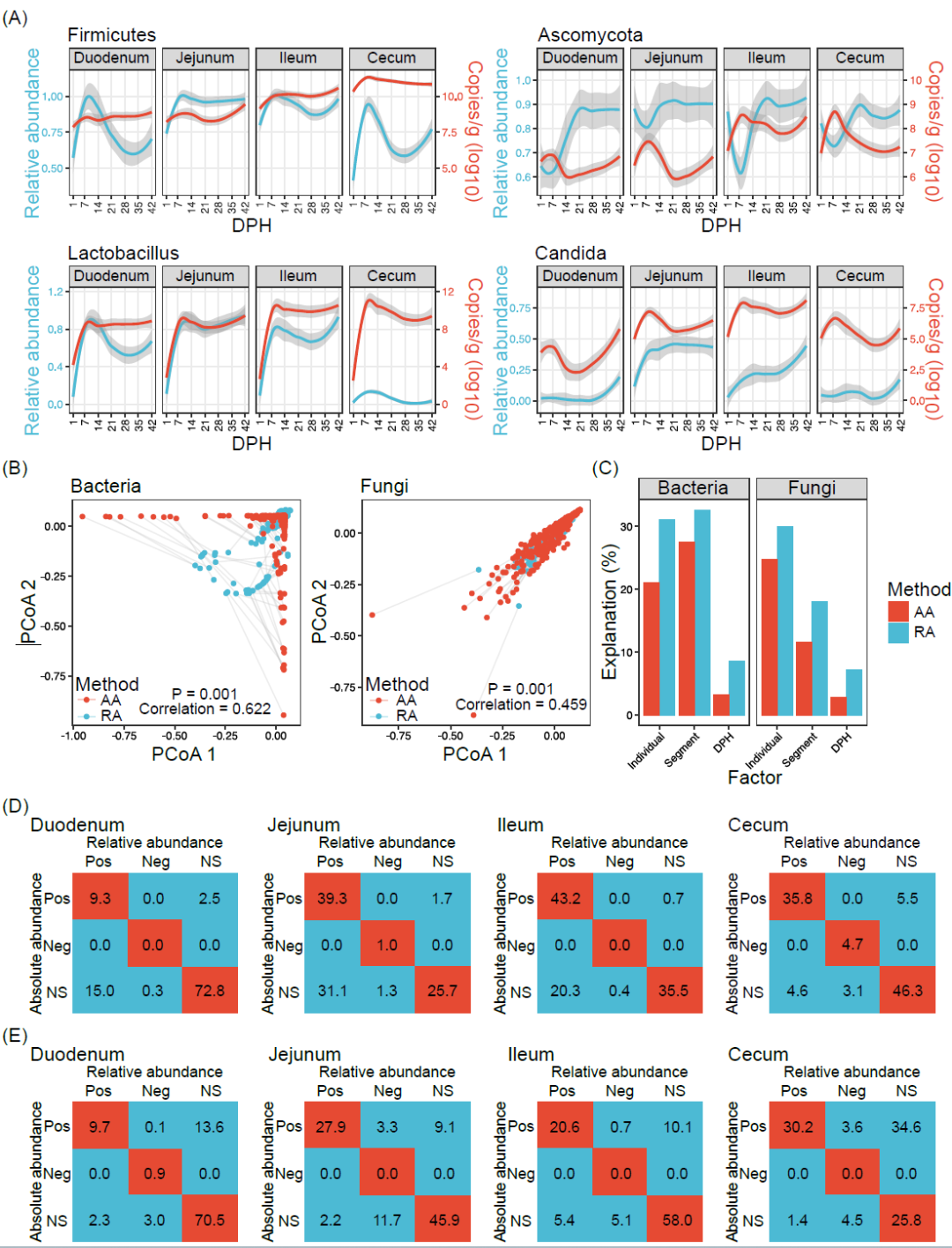
图 2. 定量微生物组和相对定量微生物组方法在解析鸡肠道微生物的差异
(A)主要的门(厚壁菌门和子囊菌门)和主要的属(乳酸菌属和念珠菌属)的绝对丰度和相对丰度的变化。红线:绝对丰度;蓝线:相对丰度。(B)细菌和真菌的绝对丰度和相对丰度之间的相关性的普氏分析。红点:细菌或真菌的绝对丰度;蓝点:细菌或真菌的相对丰度。(C) 由个体、肠段和日龄等因素解释的肠道微生物群变异百分比。基于绝对丰度和相对丰度的细菌(D)和真菌(E)物种间相关性结果的一致性分析。Spearman相关性结果中调整后p值小于0.05被认为是显著相关的。单元格中数字是不同类别中每对相关性的比率。AA,绝对丰度;RA,相对丰度;DPH,孵化后天数;Neg,负相关;Pos,正相关;NS,相关性不显著;PCoA:主坐标分析;QMP,定量微生物组分析;RMP,相对定量微生物组分析。
确定性和随机过程塑造肠段特异的群落类型
为了进一步研究使用定量微生物组方法下肉鸡肠道微生物的动态变化,我们首先使用Dirichlet多项式混合模型(DMM)对不同肠段的群落类型进行分类。我们将细菌菌群和真菌菌群分别划分成了五种和四种不同的群落类型。乳酸菌属、链球菌属和粪杆菌属主导了细菌群落类型,四孢镰刀菌属、扁囊孢属、镰刀菌属和念珠菌属主导了真菌群落类型(补充材料图S5A、B)。这些群落类型在微生物数量、香农指数和微生物组成方面有显著差异(p < 0.05,补充材料图S5C-H),并具有不同的时空分布特征(图3A,B)。在1日龄时,所有肠段中只有细菌群落1型和真菌群落4型存在。从4日龄开始,细菌群落4型和5型在十二指肠中较常见,1型和2型则在空肠和回肠中很普遍,而3型则在盲肠中占据主导地位。对于真菌菌群,2型在空肠更常见,3型在盲肠出现的频率更高。这些结果表明,鸡肠道微生物在孵化后就迅速形成肠段特异的群落类型。
肠段特异的微生物分布促使我们进一步研究前肠微生物对后肠微生物的影响。我们使用FEAST来研究后三个肠段(空肠、回肠和盲肠)的微生物来源情况。我们发现从十二指肠到空肠及从回肠到盲肠的微生物传递有限,只有空肠微生物贡献大约一半的回肠微生物(图3C)。我们使用生态学模型来研究各肠段形成特定群落类型的内部驱动力。结果表明,在细菌菌群中确定性过程(56.7%)比随机过程(43.3%)重要,而在真菌菌群中随机过程(55.1%)比确定性过程(44.9%)重要(图3D)。均匀分散(细菌32.7%,真菌50.3%)和同质选择(细菌56.7%,真菌44.0%)主导细菌菌群和真菌菌群结构。细菌菌群中高比例的同质选择可能导致每个肠段中拥有相似且稳定的菌群结构。除了空肠真菌菌群外,真菌菌群在均匀分散下处于不可预测和无序的群落状态。在最近的一项研究中也发现鸡肠道内真菌菌群的高度动态变化。
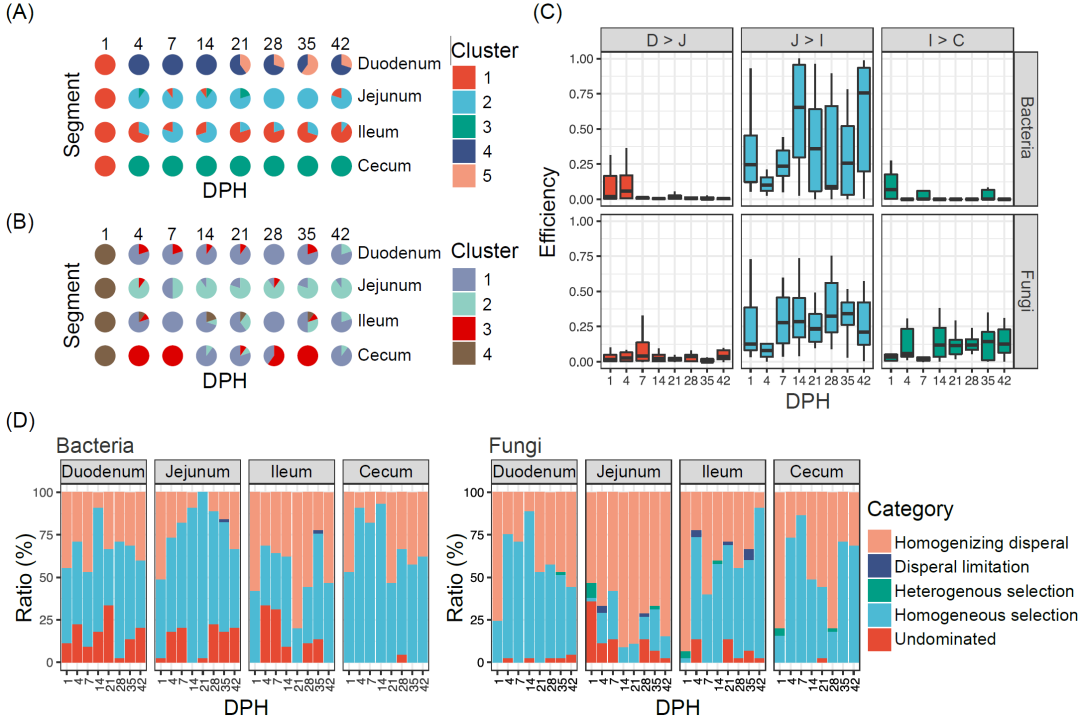
图 3. 细菌和真菌菌群的聚类与肠段和选择压力有关
细菌菌群(A)和真菌菌群(B)的DMM聚类。不同颜色代表不同的DMM类型。(C)前面的肠微生物对邻近的后面的肠微生物的相对贡献。(D)不同的生态过程的重要性(异质选择:βNTI < -2,同质选择:βNTI > 2,扩散限制:|βNTI| < 2且RCBray > 0.95,均匀分散:|βNTI| < 2且RCBray < -0.95,以及无支配:|βNTI| < 2且|RCBray| < 0.95)。βNTI,β-最近的分类群指数;DMM,Dirichlet 多项式混合模型;DPH,孵化后天数;RCBray,基于Bray-Curtis的Raup-Crick指数。
鸡肠道中微生物定植能力不同
我们研究了42天内细菌和真菌的定殖时序。肉鸡肠道微生物在不同肠段的定植时序在补充材料图S6(细菌)和图S7(真菌)中。四个肠段按照定植时间划分的模式数量从71到168不等(补充材料表S6)。大多数模式在整个42天周期中的时间点是以“混合”的形式存在,这表明很大一部分微生物在不同个体中并不能稳定存在。然而,在特定肠段能够发现稳定的定植模式。例如,在盲肠中存在两种定植模式含有属的数量最多(21和14,补充材料图S6)。这两种模式的细菌能够快速定植(分别在4日龄和7日龄开始),并成为肠道微生物中稳定组成部分。我们进一步将所有呈现出规律变化的模式(n = 23)分为三类:“定植”、 “消失”和“核心”(图4A和补充材料表S7)。“定植”这类一共包含15个模式(107个属),这些模式的微生物在孵化伊始没有定植,但在后来时间中出现。值得注意的是,该类别中的两个属和五个真菌属在四个肠道段中都存在相同的模式(1日龄时开始定植,此后稳定存在,图4B和补充材料表S8)。7个模式的24个属被划分到“消失”的类别,它们在1日龄时出现,在随后时间点消失。值得注意的是,真菌(n = 22)比细菌(n = 2)更容易出现在这种模式,这表明一些细菌在定植过程中具有相对竞争优势。没有发现在四个肠段中都属于这个模式的微生物(图4C和补充材料表S9)。一个包含18个属的模式属于“核心”类别,即在整个42日龄中都存在。这18个属的绝对丰度在整个时间段中在特定的肠段或整个肠道中数量相对恒定(补充材料图S8),这表明具有高度的稳健性和自我调整能力。在这18个属中,有四个真菌属在所有四个肠段都存在(图4D和补充材料表S10),这表明这些真菌比细菌和其他真菌拥有更强的适应能力。
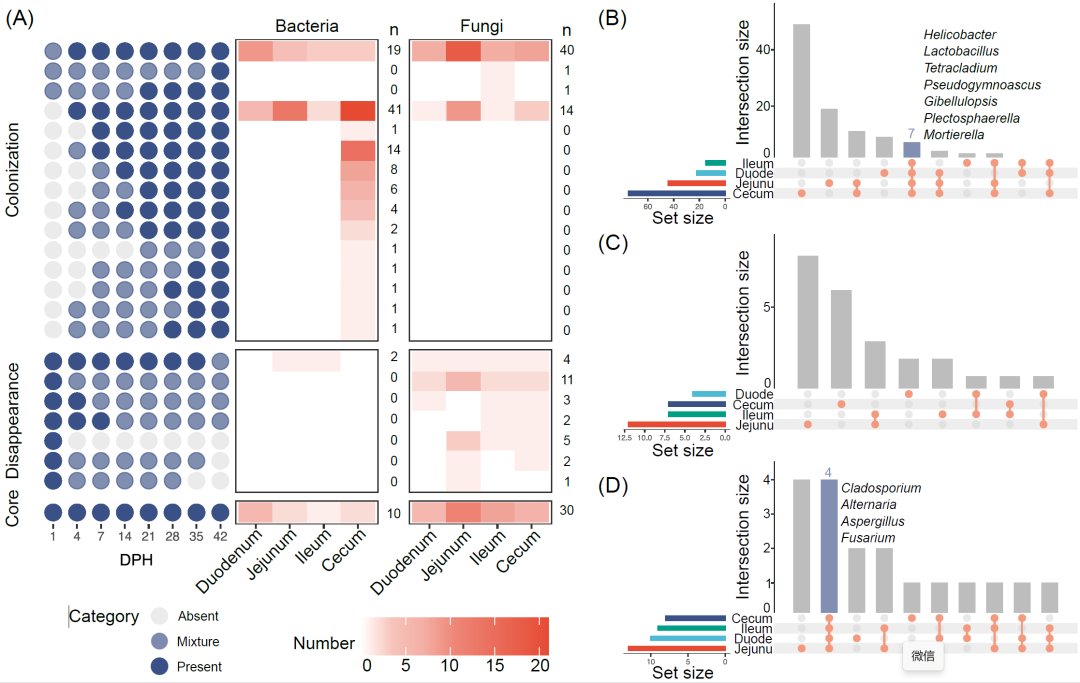
图 4. 四个肠段中微生物属水平发育模式和变化
(A)四个肠段中发育模式总结成三个分类(核心、消失和定植)。不同颜色代表不同的DMM类型。深蓝色的点代表该微生物存在(n ≥ 9);灰色的代表该时间点没有该微生物;浅蓝色的点代表微生物存在状态介于“存在”和“不存在”之间。每个格子颜色代表不同模式的数量。UpSetR图展示定植(B)、消失(C)和核心(D)分类的交集。DPH,孵化后天数。
共现网络揭示鸡肠道内微生物之间更多的正相关关系
我们通过整合八个采样点的属水平相互关系,构建四个肠段的微生物共现网络(补充材料图S9)。四个网络的拓扑学特征是不一样的(图5A和补充材料表S11)。盲肠微生物共现网络的顶点数(n = 274)和边数(n = 19,626)比其他三个肠段共现网络多。空肠微生物共现网络的聚类系数(0.79)和盲肠微生物共现网络的聚类系数(0.81)高于其它两个肠段,空肠微生物群平均介数较低(36.0)。十二指肠和回肠微生物共现网络呈现出较高的平均分离度(分别为1.74和1.79)。这些结果表明,盲肠微生物呈现出更复杂的微生物网络结构,具有更多的顶点数、边数、聚类系数、平均介数和较低平均分离度值。前三个肠段拥有相对较高比例(从52.1%到92.8%)共有边(至少存在于两个肠段),但盲肠包含了更多数量(92.8%)的特异边(只存在于一个肠段,图5B),这表明随着微生物丰度和多样性增加,许多新的微生物相互关系在盲肠中建立。
接下来,我们研究了四个共现网络中的关键微生物,即高度连接的微生物。图5C显示了每个网络中度数最高的10个顶点(共计34个属)。[Ruminococcus] torques group、瘤胃梭菌属9、链格孢属/f_Nectriaceae和瘤胃梭菌属5分别是十二指肠、空肠、回肠和盲肠的关键微生物,Erysipelatoclostridium、Sellimonas、丁酸球菌和瘤胃梭菌属9是一个以上共现网络的关键微生物。在这34个属中,29个属属于“定植”模式,4个属属于我们定义的“核心”模式(补充材料表S12)。值得注意的是,在这些关键微生物当中有8个属属于瘤胃球菌科,突出了它们在鸡肠道中的重要作用。我们进一步分析了每个网络中各个属的正相关关系和负相关关系。有趣的是,绝大多数的属,包括所有的关键微生物,与其他属的正相关关系数量多于负相关关系数量(图5D)。只有少数属与其它微生物负相关关系数量比正相关关系数量多,包括红酵母属、埃希氏菌属和志贺氏菌属。
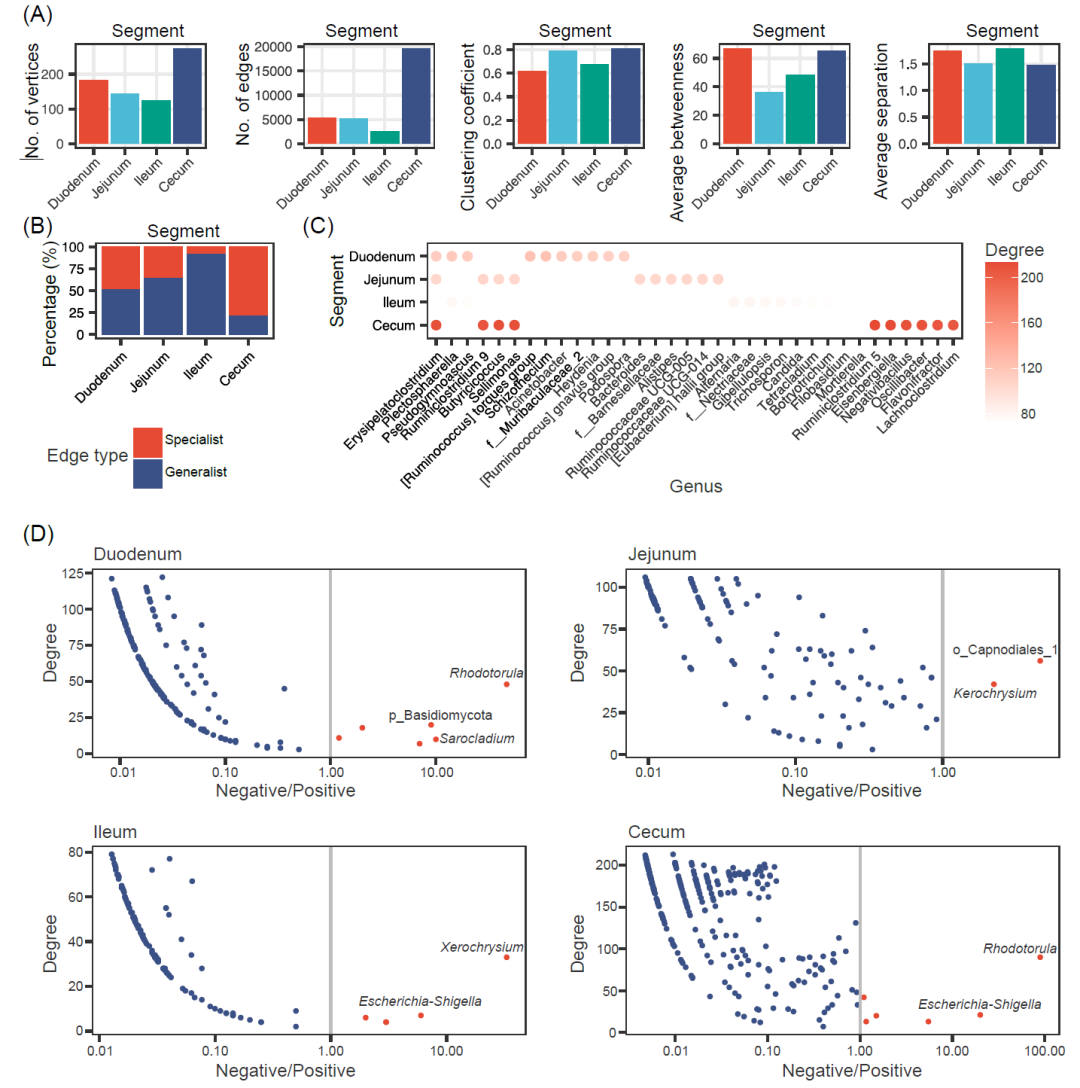
图 5. 四个肠段肠道微生物共现网络
(A)基于微生物绝对丰度推断出的微生物共现网络的拓扑学特性。(B)四个微生物共现网络中共有边和特异边的比例。四个微生物共现网络中共有边和特异边的比例。(C)四个肠道段中的前10个关键微生物。每个点的颜色代表每个微生物的平均度。空白位置表示该微生物不是一个关键微生物。(D)负相关关系数量和正相关关系数量比例的对数转换(log10)与微生物度的值的散点图。在不同肠段(十二指肠、空肠、回肠和盲肠)中每个微生物的负相关关系和正相关关系数量比率的散点图。红点:负相关关系数量多于正相关关系数量;蓝点:正相关关系数量多于负相关关系数量。
血清代谢物丰度伴随着鸡的发育而变化
然后,我们进行了非靶向代谢组学的时序研究,以了解肉鸡血清中代谢物在生长过程中的变化。首先,使用OPLS-DA对样品进行聚类,形成三个互相分离的组(1日龄组,4-21日龄组和28-42日龄组)。新孵化小鸡血清代谢物与其他两个生长阶段不同(FDR < 0.05),并且从21日龄到28日龄发生了变化(图6A,B和补充材料表S13)。为确定不同时间点之间分离有显著贡献的代谢物,我们选择了预测中VIP值大于1.5作为代谢物的阈值(图6C)。共有30个代谢物符合这一标准,2-heptyl-4,5-dimethylthiazole、lycimumoside VIII和lyciumoside III是贡献度最大的三个。
为确定随时间变化的代谢物,我们使用一般线性模型进行分析。在1109种代谢物中共有641种与时间有关(一般线性模型,FDR < 0.05),并形成六个不同的簇(图6D和补充材料表S14)。这些簇中代谢物的丰度遵循不同的变化轨迹(图6E):簇1(22个代谢物)和簇3(143个代谢物)的丰度随时间增加;簇6(37个代谢物)的丰度随时间减少;簇2(318个代谢物)和簇4(24个代谢物)代谢物在1 日龄后表现出急剧的丰度变化,分别从低到高和从高到低。簇5(97个代谢物)的丰度先上升后下降。六个簇在代谢物组成上有差异:簇1有较高比例的苯类化合物;簇2和簇3包含更多的脂类和类脂分子;有机酸和衍生物在簇4中更多,有机氧化合物在簇6中占比高(图6F)。我们分析代谢物丰度随时间变化后发现丰度变化最大的30个代谢物中,有14个代谢物来自于簇1,其丰度都随时间而增加(图6G)。
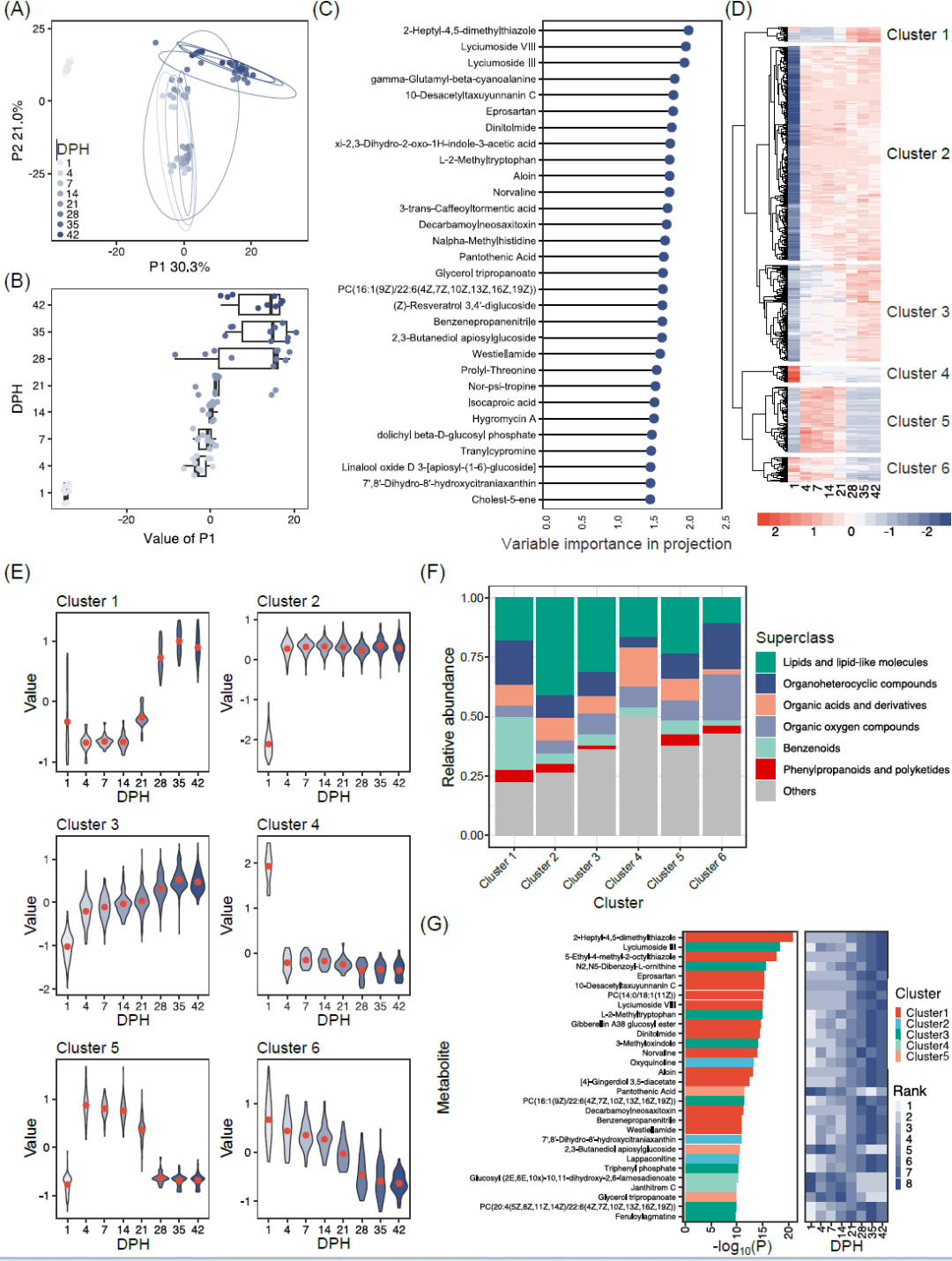
图 6. 肉鸡生长过程中血清代谢物的变化
(A)不同时间点鸡血清代谢谱的OPLS-DA图。(B)不同时间点的OPLS-DA结果的P1值。P1能够解释组间30.3%的变化。(C)按VIP值排列的代谢物重要性。(D)随时间显著改变的代谢物丰度热图(一般线性模型,FDR < 0.05)。根据分层聚类,将代谢物分为六个簇。(E)六个簇的代谢物丰度随时间变化趋势。(F)每个簇代谢通路分析总结。(G)根据一般线性模型结果,前30随着时间的推移显著变化的代谢物。条形图的颜色代表簇的来源。单元格颜色反映了不同时间点的丰度排名。GLM,一般线性模型;FDR,错误发现率;OPLS-DA,正交部分最小二乘法判别分析;VIP,投影中的变量重要性。
基于定量微生物组建立微生物和鸡血清代谢物的关联
为探究鸡肠道微生物动态变化与血清代谢物间的相关性,我们分析了不同肠段微生物对血清代谢物变化的解释度。我们发现,来自不同肠道段的细菌菌群和真菌菌群与血清代谢物显著相关(p < 0.001,protest检验,补充材料表S15)。进一步比较残差结果显示,前肠微生物和血清代谢物之间的残差低于后肠微生物和血清代谢物之间的残差(图7A和补充材料表S16),表明虽然鸡前肠(十二指肠和空肠)微生物的多样性很低,但这些肠段的微生物可能对宿主的代谢物影响更大。这些结果突出了肠段差异影响微生物菌群和血清代谢物之间关联。
我们使用mmvec来预测微生物-代谢物之间的关联。基于不同肠段共同出现概率,通过选择条件概率最高的1%的微生物-代谢物关联生成了包含180个属和560种代谢物的共现条件概率热图(补充材料图S10)。我们研究了它们在四个肠段中的分布情况,发现肠道微生物群与簇1、簇3和簇5中代谢物的关系更密切(图7B)。空肠中真菌菌群(2.38%)与簇1中代谢物关系最密切,其次是十二指肠中细菌菌群(1.54%)和真菌菌群(1.41%),分别与簇1和簇5中的代谢物关系密切。结果还显示,在四个肠段中,来自子囊菌门和厚壁菌门的属与其他门相比,其与簇2和簇3中代谢物关联更紧密(补充材料表S17和图S11)。
为了进一步探索微生物-代谢物的相互关系,我们整合四个肠段分别建立的所有可能微生物-代谢物相互关系,构建了一个新的共现网络(图7C)。结果表明,更多的来自子囊菌门(n = 68)和厚壁菌门(n = 58)的属与代谢物关联密切,不整小球囊菌属、[Ruminococcus] torques group、p__Ascomycota、丁酸球菌属、腐质霉属、o__Hyprcreales_2和乳酸杆菌属是微生物-代谢物相互关系中最重要的微生物(图7C和补充材料表S18)。与微生物相关的代谢物主要来自于脂质和类脂质分子(n = 184),其次是有机杂环化合物(n = 57)和有机酸及其衍生物(n = 55,补充材料表S19)。在184个微生物相关的脂类和类脂分子中,分别有62个、51个和42个代谢物来自脂肪酰基、前醇脂和甘油磷脂类,更多的代谢物属于它们的亚类甘油磷脂(n = 28)、脂肪酸酯(n = 14)、三萜类(n = 14)和萜类糖苷(n = 14,补充材料表S20)。有机杂环化合物类别中代谢物分布在类或亚类水平上,更多化合物来自唑类(n = 7),吲哚类及其衍生物(n = 6),吡啶类及其衍生物(n = 5)。大多数微生物相关的有机酸及其衍生物来自羧酸和衍生物(n = 45),主要是氨基酸、肽及类似物(n = 43),包括必需氨基酸(L-赖氨酸)、非必需氨基酸(L-酪氨酸)、非蛋白质氨基酸(正缬氨酸)和二肽(如谷氨酰-组氨酸和谷氨酰-异亮氨酸)。
然后我们使用MetOrigin平台预测这些代谢物可能来源。我们确定了11种肠道微生物来源的代谢物,以及16种共同来自宿主和肠道微生物的代谢物,包括L-赖氨酸、L-酪氨酸等(补充材料表S21)。功能富集分析表明,这些代谢物主要参与氨基酸代谢(赖氨酸降解和缬氨酸、亮氨酸和异亮氨酸降解)和脂质代谢(鞘脂代谢和甘油磷脂代谢,补充材料表S22)。
总之,基于鸡肠道微生物发育的绝对丰度情况确立肠道微生物和肉鸡血清代谢物参与代谢之间的密切联系。
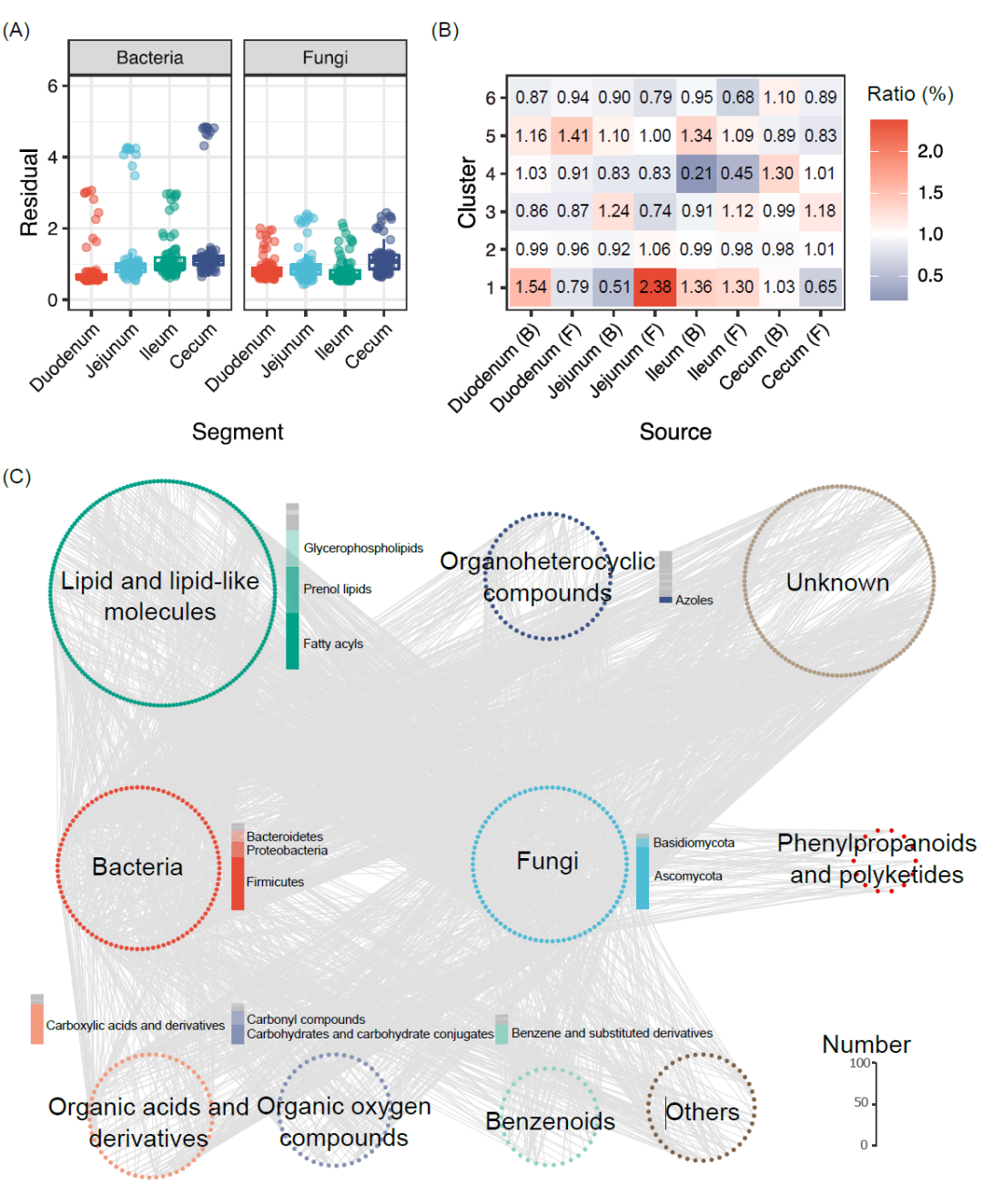
图 7. 微生物-代谢物的相互关联
(A)绝对丰度下不同肠段微生物-代谢物关联残差的差异。(B)所有微生物-代谢物相互关联中前1%共现可能性的比率。红色单元格代表代谢物和微生物之间存在更多的相互关联;蓝色单元格代表代谢物和微生物之间存在较少的相互关联。(C) 使用mmvec从四个肠段发现的微生物-代谢物相互关联共现网络。条代表不同亚类代谢物的数量。mmvec, 微生物体-代谢物向量。
讨 论
本研究利用定量微生物组方法研究肉鸡肠道微生物(细菌菌群和真菌菌群)的发育轨迹,并将42天的生产周期不同肠段微生物动态变化与鸡血清代谢物联系起来。据我们所知,本研究是第一个使用高通量测序辅以定量微生物组方法研究鸡肠道内细菌菌群和真菌菌群变化的研究。如其他微生物组学研究所示,绝对定量在最大限度地减少相对定量分析所引入组成效应影响方面具有优势。
我们发现,在鸡孵化后前7天,细菌和真菌迅速定植于肠道中。四个肠段中的早期定植的细菌主要是需氧菌和兼性厌氧菌,如链球菌属、埃希氏菌属和志贺氏菌属。这些细菌甚至在4日龄之前就被厌氧菌所取代,这可能是由于肠道内氧气迅速耗竭。尽管肠道微生物多样性在7日龄时达到稳定状态,但鸡肠道微生物组成和功能仍可能发生改变。例如,盲肠中拟杆菌门(一类能够降解复杂的碳水化合物的微生物)在14日龄时开始增殖。真菌菌群多样性随时空变化较小。然而,从4 日龄开始,空肠和回肠中出现大量念珠菌属值得更多关注。与细菌和真菌相比,鸡肠道中古菌的检出率和丰度都很低。我们以前研究也发现,在799个鸡肠道样本得到的1978个种水平组装基因组中,只有8个种是古菌。粪便和苍蝇是鸡肠道古菌的潜在来源。鸡肠道内古菌检出率低可能是由于现代养殖系统提供了更好的卫生条件。
以前研究表明,定量微生物组方法所揭示的人类肠道和自然环境中微生物菌群组成与相对定量微生物组方法所揭示的组成有显著差异。在本研究中,我们也发现鸡肠道微生物基于定量微生物组方法和相对定量微生物组方法得到结果的不一致。首先,定量微生物组结果表明,鸡肠道不同肠段微生物绝对数量并没有像相对定量结果所揭示的那样有很大变化,而且与相对定量微生物组结果相比,其绝对丰度在一段时间内相对稳定(图2A)。这一结果表明,相对定量微生物组方法得到的鸡肠道微生物结果与定量微生物组结果有偏差。第二,相对定量微生物组方法高估了微生物群落结构的变化及其影响因素。第三,相对定量微生物组方法遗漏了绝对定量中发现的许多微生物间的关联,特别是在真菌菌群。这些结果表明,定量微生物组方法是必要的,并将提供新线索来理解鸡肠道微生物的变化及微生物与宿主之间的关联。
揭示鸡肠道微生物在整个生产期间的建立和演替可能为有效干预肠道微生物,改善鸡健康状态和生产性能提供机遇。群落类型(或肠型)被定义为肠道微生物组成的标志物,其在样本中的变化经常被用来探究肠道微生物的变化。通过分析群落类型,我们证明了鸡肠道微生物(包括细菌和真菌)随着宿主发育表现出肠段特异性。本研究和以前研究中微生物溯源分析结果表明,后面肠段的一小部分微生物(10%至30%)来自相邻前面肠段。鸡肠段特异微生物的产生可能是由于微生物的条件性定植:(1)宿主选择压力,如不同肠段具有不同的物理结构、条件和功能;(2)微生物的适应性和生存能力;(3)微生物菌群内的竞争和排斥。从生态学角度来看,这种类型的定植可能是一个确定性过程。然而,确定性和随机过程共同影响着微生物的建立。我们发现,两种过程都影响着鸡肠道微生物的定植。确定性过程主导了细菌菌群的建立,而随机过程主导了真菌菌群的建立。这种差异可能解释细菌菌群比真菌菌群为什么会表现出更多不同的群落类型和建立过程。
除了肠段特异微生物外,我们还在鸡肠道中发现了阶段特异微生物。阶段特异微生物被证明对人类和猪的发育和健康至关重要。为了确定阶段特异性微生物,我们将微生物菌群的发育模式分为三类:“定植”、“消失 ”和“核心”。大多数细菌和许多真菌都属于定植模式,特别是在鸡盲肠中,包括许多有益微生物如乳酸杆菌、拟杆菌属和瘤胃球菌科的成员,以及几个很少被关注的真菌,如四孢镰刀菌属、Leohumicola、 Pseudogymnoascus和Mortierella。尽管与细菌相比,更多真菌属于“消失”模式,而细菌可能不具有竞争优势,但更多真菌在整个生产周期持续存在(“核心”)。在属于“核心”类别的18个属中,有4种真菌均存在于四个肠段。在后续分析中,我们发现真菌和鸡血清代谢物之间联系密切。这些结果表明,真菌菌群在鸡发育和健康中作用被忽视。最近的微生物组研究中,肠道真菌菌群引起越来越多的关注。例如,肠道真菌菌群与宿主免疫细胞有密切的关系,共生真菌菌群的破坏会影响局部和外周的免疫反应,并影响小鼠相关疾病状态。
微生物共现网络被广泛用于研究肠道微生物中微生物间关系,网络中微生物相互作用与微生物竞争强度不同或生态位分化有关。我们研究表明,与小肠相比,鸡盲肠微生物共现网络最为复杂,十二指肠中微生物相互作用比空肠和回肠中相互作用复杂。在猪肠道微生物中也发现类似的现象。尽管十二指肠中微生物的绝对数量低,但微生物多样性高于空肠和回肠,这可能解释了为什么呈现出更多微生物相互关系。我们推断,十二指肠中较高微生物多样性可能是由于它直接从胃中接收和积累了不同微生物。在受到宿主选择压力后,某些微生物在空肠和回肠的数量增加,但微生物间相互作用减弱,然后微生物进入盲肠发酵增殖。
微生物相互关系网络中关键微生物被定义为高度连接的微生物,无论其丰度如何,都可能对微生物结构和功能产生较大影响。我们发现,四个网络中几乎所有关键微生物都呈现出有规律的定植模式,即“定植”和/或“核心”,这表明具有竞争优势的微生物在稳定微生物相互作用方面起到了关键作用。所有的关键微生物与网络中其他属表现出更多正相关关系,而不是负相关关系。瘤胃球菌科是一个典型的代表。属于瘤胃球菌科的8个属是关键微生物,其定植模式为“定植”。瘤胃球菌科是人和动物肠道中常见的微生物,它们能分解复杂的碳水化合物,产生有益代谢产物,如短链脂肪酸,能够维持肠上皮细胞功能和形态,调节肠道微生物平衡。瘤胃球菌科的物种能通过互养与其他微生物保持密切联系。我们研究结果证实瘤胃球菌科细菌作为关键微生物以及它们与鸡肠道中与其他微生物的正相关关系。然而,我们对瘤胃球菌科在鸡的健康、发育和生产性能方面功能的相关研究还是空白,值得在未来给予更多关注。
我们发现,尽管前肠中真菌丰度较低,但它们与鸡血清代谢物间联系密切。在以前研究中也观察到前肠微生物与鸡代谢和生长间的关联。这一结果可能是由于微生物代谢物直接吸收或微生物对小肠中膳食营养吸收的间接影响。人类小肠微生物失调与功能性胃肠道疾病和慢性肝病有关。因此,今后应加强前肠微生物在鸡生长发育中的作用和内在机制的相关研究。
肠道微生物是决定宿主血清代谢状况的一个重要因素。我们结果表明,来自子囊菌门和厚壁菌门的微生物与血清代谢物之间存在更多关联(图7C)。子囊菌门和厚壁菌门分别是鸡肠道真菌和细菌中的优势菌门,我们的结果证实它们对鸡生理和代谢,特别是脂质和氨基酸代谢的重要影响。
越来越多的证据表明,微生物改变了循环系统中脂质浓度,并影响宿主脂质平衡。我们发现,鸡肠道微生物主要与属于脂肪酰基、前醇脂和甘油磷脂的化合物有关。对于脂肪酰基类,与花生四烯酸关联的肠道微生物数量最多。这一结果也得到了相关研究结果的支持,改变微生物菌群会明显扰乱花生四烯酸代谢。花生四烯酸可通过代谢产生生物活性脂肪酸介质,是与鸡肉风味有关的关键物质。对于甘油磷脂,属于甘油磷脂的28种代谢物与鸡肠道微生物群有关,这与小鼠的结果相似,即肠道微生物群通过改变甘油磷脂的酰基链谱,包括磷脂酰胆碱、乙醇胺和肌醇,改变小鼠脂质特征。我们在以前研究中还发现,宿主遗传依赖的微生物与鸡肌肉脂质代谢高度相关。
肠道微生物能够从头合成必需氨基酸,而这些氨基酸与宿主体内氨基酸平衡有关。我们发现,L-赖氨酸是肉鸡玉米-豆粕日粮中第二限制氨基酸,其血液中含量与肠道微生物密切相关。这一结果与前人研究结果一致,即肠道微生物产生的L-赖氨酸中大约75%在小肠中被吸收。我们研究还表明,L-酪氨酸是一种非必需氨基酸且与肠道微生物有关,其在肠道中含量也高于其他组织。我们还发现肠道微生物和肽以及几种氨基酸类似物间的联系,如加巴喷丁和L-正缬氨酸。二肽在保护肠道环境免受氧化应激和调节免疫系统方面发挥重要作用。加巴喷丁是γ-氨基丁酸类似物,能够增加体重和采食量提高生长性能。L-正缬氨酸促进组织再生和肌肉生长。
综上所述,我们基于定量微生物组方法分析肉鸡肠道细菌菌群和真菌菌群发育,揭示肠道微生物和鸡血清代谢物间潜在联系。结果表明,相对定量结果对鸡肠道微生物发育和微生物间相互关系解读有一定偏差,并利用定量微生物组方法揭示肠段特异的微生物群落和阶段特异的微生物。我们还发现前肠微生物和真菌菌群在鸡脂质和氨基酸代谢中的重要性。这个研究全面展示鸡生长过程中肠道微生物发育的动态变化,并提供关于肠道微生物对鸡代谢影响。我们也要说明的是,肠道微生物和鸡血清代谢物间关联结果只是基于关联分析得到;未来实验验证工作去证实我们的发现是十分必要的。
方 法
动物饲养和样品采集
同一天孵化的200只雄性爱拔益加雏鸡在同一个养鸡场(中国涿州)进行饲养。肉鸡日粮配方是根据中国鸡饲养标准(NY/T33-2004,补充材料表S1)确定。肉鸡被分为10个重复。根据实验设计,对每一个重复进行了8个时间点的采样,每次随机抽取一只鸡。收集全血和四个肠段(十二指肠、空肠、回肠和盲肠)内容物。更多细节在补充材料中。
DNA提取和定量
微生物基因组提取是根据以前描述的方法进行。由于机械破碎和反复震动可以提高基因组提取效率,我们通过添加微珠并进行三轮机械破碎来提高破碎效率。使用QIAamp Stool DNA mini Kit(货号:51504,Qiagen)提取每个肠段内容物中微生物基因组DNA。在提取基因组DNA过程中记录提取用样品重量(补充材料表S2)。
对于微生物数量,我们首先构建了一个以10为对数的标准曲线,该曲线的范围从103到108拷贝,选择16S或ITS rRNA基因拷贝数已知的参考基因组(大肠杆菌,毕赤酵母)用细菌、古菌和真菌的定量。
细菌、古菌和真菌的定量是通过实时荧光定量PCR方法进行,使用ABI 7500 PCR系统(Applied Biosystems),反应体积为20 μL,含有10 μL的TB Green ® Premix Ex TaqTM(货号:RR420A,Takara)。用于定量细菌数量的引物是5'-TCCTACGGGAGGCAGCAGT-3'(正向)和5'-GGACTACCAGGGTATCTAATCCTGTT-3'(反向)。用于定量真菌的引物是5'-CTTGGTCATTTAGAGGAAGTAA-3'(正向)和5'- TCCTCCGCTTATTGATATGC-3'(反向)。更多细节在补充材料中。
扩增子测序和数据处理
为进行不同界的扩增子测序,样品被分成两份,每份都用特定引物对进行扩增:引物V3-V4(正向5’-CCTACGGGNBGCASCAG-3’和反向 5’-GACTACNVGGGTATCTAATCC-3')用于原核生物(细菌和古菌),或引物ITS2(正向5’-GTGARTCATCGAATCTTT-3’和反向5’-GATATGCTTAAGTTCAGCGGGT-3’)用于真核生物。构建好的文库在Illumina HiSeq平台上进行测序(2 × 250 bp,补充材料表S3)。
使用QIIME2对原始测序文件进行质量过滤和物种分析。我们使用DADA2将质量过滤后序列拼接成扩增子序列变体(ASV)。基于SILVA数据库和UNITE数据库对ASV进行物种注释。使用R包vegan计算香农指数和基于Bray-Curtis距离的主坐标分析(PCoA)。α多样性和β多样性的统计检验分别使用Kruskal-Wallis检验/Wilcoxon秩和检验和PERMANOVA进行。基于相对丰度和绝对丰度微生物组间的Procrustes相关系数和p值是使用vegan中的“protest”计算得到。更多细节在补充材料中。
肠型聚类和肠道微生物建立的随机性
使用DMM模型将样本划分到不同群落类型,并根据对数转换后绝对丰度对样本进行分类。FEAST算法用来对微生物进行溯源。从空肠开始,每个采样点都被确定为终点,所有其他样本被视为潜在来源。在本研究中,只考虑来自相邻肠道前段微生物。为评估微生物组建立中确定性过程和随机过程的重要性,使用β-最近分类群指数(βNTI)和基于Bray-Curtis的Raup-Crick指数(RCBray)值进行分析,包括异质选择、同质选择、扩散限制、均匀分散和未支配。随机过程包括三个过程(均匀分散、扩散限制和未支配),确定性过程包括两个部分(异质选择和同质选择)。βNTI和RCBray的数值是用R包iCAMP计算得到。关于肠型聚类和肠道微生物建立的随机性更多细节在补充材料中。
微生物共现网络分析
微生物间共现网络是使用R包ppcor在不同的肠段中使用Spearman偏相关构建而成。为了降低噪音和假阳性比例,网络分析仅纳入至少在30%样本中都存在的属。在每个微生物网络中使用FDR过滤掉调整后p值超过0.05的相关关系。使用R包igraph进行网络拓扑学特征分析。只出现在一个子网络中的边定义为特异边,而出现在一个以上子网络中的边被定义是通用边。关键微生物是高度关联的微生物,对微生物组结构和功能有较大影响。更多细节在补充材料中。
鸡血清代谢组学分析和数据处理
我们进行非靶向代谢组学分析鸡血清中代谢物。为了分析鸡血清中代谢物,我们使用了Agilent 1200系列高效液相色谱系统(Agilent Technologies, CA)来分离代谢物。安捷伦Mass Hunter工作站软件被用来进行数据分析和代谢物鉴定。下游分析基于METLIN和HMDB数据库注释结果,进行两步过滤处理:(1)保留了大于30%样品中存在的代谢物;(2)过滤掉未知的代谢物。共有1109种代谢物用于后续分析。
使用R包ropls中OPLS-DA对不同时间点血清代谢物进行了分析,并计算代谢物的VIP值。我们使用R包pheatmap将代谢物聚类为六个簇。使用MetaboAnalyst 5.0(http://www.metaboanalyst.ca)对每个簇中代谢物进行代谢通路分析。相关代谢物的类别和功能富集分析使用MetOrigin平台进行。更多细节在补充材料中。
微生物-代谢物的相互关联
为了研究微生物和代谢组之间的关联,我们分析了基于相对/绝对丰度的微生物和代谢谱之间的对称Procrustes相关系数。p值是使用vegan中的“protest”计算得到。相关性残差是由R包vegan中“procrustes”函数计算得到。使用mmvec对来自不同肠段微生物和代谢谱进行分析,根据神经网络预测共同出现概率来确定微生物-代谢物间关联。条件概率最高的1%微生物-代谢物关联被认为是阳性结果。
统计分析和可视化
上面没有提到的相关性是用R函数“cor.test”计算得到。当同时考虑多个假设检验时,调整p值以控制FDR。使用R包UpSetR分析不同集合的交集。使用Gephi对共现网络进行可视化。微生物-代谢物关联热图是用R包ComplexHeatmap进行可视化。本研究中大多数可视化是用R包ggplot2和patchwork完成。在鸡生长发育过程中,使用一般线性模型对代谢物时序数据进行分析。
代码和数据可用性:
所有测序数据都已存入NCBI的SRA数据库(PRJNA817429,https://www.ncbi.nlm.nih.gov/sra/?term=PRJNA817429)和国家微生物学数据中心(NMDC10018360, https://nmdc.cn/resource/genomics/project/detail/NMDC10018360)。项目使用脚本存放于GitHub(https://github.com/yuqing-feng/scripts_for_iMeta_2023_11.git)。补充材料(文本、图、表、中文翻译版本或视频)可从线上获取。
引文格式:
Yuqing Feng, Meihong Zhang, Yan Liu, Xinyue Yang, Fuxiao Wei, Xiaolu Jin, Dan Liu, Yuming Guo, Yongfei Hu. 2023. Quantitative microbiome profiling reveals the developmental trajectory of the chicken gut microbiome and its connection to host metabolism. iMeta e105. https://doi.org/10.1002/imt2.105
作者简介

封雨晴(第一作者)
● 中国农业大学动物科学技术学院副教授,硕士生导师。
● 2022年10月以“优秀人才”引进中国农业大学动物科学技术学院。主要研究方向为家禽肠道微生物功能解析和微生物比较基因组学。目前,已发表SCI论文20余篇,其中以第一或通讯作者在iScience、Communications Biology、iMeta、STAR Protocols等杂志发表学术论文15篇。

胡永飞(通讯作者)
● 中国农业大学动物科学技术学院教授,博士生导师。
● 2018年6月以“杰出人才”引进中国农业大学;中国农业大学青年科学家创新团队负责人。先后主持国家和省部级项目10余项;发表SCI论文60余篇,其中以第一或通讯作者在Nature Communications、The Lancet Infectious Diseases、Microbiome等杂志发表40余篇,2篇为ESI高被引论文,单篇最高被引662次,h-index 32;发表国内核心期刊论文20余篇;申请及授权国内发明专利6项;副主编及参编著作4部,审校译著1部。中国科学院青年创新促进会第5批会员;中国生物工程学会微生物组学与技术专业委员会委员、中国微生物学会微生物组专业委员会委员。《微生物学报》《生物工程学报》《基因组学与应用生物学》编委;Antibiotics-Basel杂志、Front Microbiol杂志Guest Editor;Front Cell Infect Microbiol杂志Associate Editor;The Innovation杂志、iMeta杂志青年编委。Nature Methods、Nature Communications、Genome Biology等杂志审稿人。获畜牧兽医青年学者讲坛“优秀奖”、颐和青年“创新奖”。

呙于明(通讯作者)
● 中国农业大学动物科技学院院长、教育部人才项目特聘教授、动物营养学国家重点实验室副主任。
● 中国畜牧兽医学会副理事长(2016年10月至今)、中国畜牧兽医学会动物营养学分会理事长(2012年10月~2016年10月)、全国新饲料评审委员会副主任(2000年10月至今)、北京市畜牧兽医学会副理事长(2015年1月至今)、《中国畜牧杂志》主编(2016年1月至今)、Animal Nutrition副主编(2012年1月至今)、Asian-Australasian Journal of Animal Science编委(2008年1月至今)和Journal of Animal Science & Biotechnology编委(2010年1月至今)等。
更多推荐
(▼ 点击跳转)
iMeta | 德国国家肿瘤中心顾祖光发表复杂热图(ComplexHeatmap)可视化方法
iMeta | 浙大倪艳组MetOrigin实现代谢物溯源和肠道微生物组与代谢组整合分析

1卷1期

1卷2期

1卷3期

1卷4期

2卷1期
期刊简介
“iMeta” 是由威立、肠菌分会和本领域数百位华人科学家合作出版的开放获取期刊,主编由中科院微生物所刘双江研究员和荷兰格罗宁根大学傅静远教授担任。目的是发表原创研究、方法和综述以促进宏基因组学、微生物组和生物信息学发展。目标是发表前10%(IF > 15)的高影响力论文。期刊特色包括视频投稿、可重复分析、图片打磨、青年编委、前3年免出版费、50万用户的社交媒体宣传等。2022年2月正式创刊发行!
联系我们
iMeta主页:http://www.imeta.science
出版社:https://onlinelibrary.wiley.com/journal/2770596x
投稿:https://mc.manuscriptcentral.com/imeta
邮箱:office@imeta.science


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









