







SNPs人群纯合/杂合度计算差异记录
摘要
多种应用下需要用到SNP人群纯合/杂合度计算作为系数或者先验,例如同源交叉污染定量、器官移植dd-cfDNA定量、遗传发育研究等。由于数据库之间存在差异,多个数据库只保留如人群AF之类的指标,并不会给出真实的位点人群纯合/杂合度(即每个位点有多少比例的人是纯合、多少比例是杂合)。基于此,本文进行人群AF(allele frequency)间接推导与真实人群纯合/杂合比例计算之间的差异探索。
数据库
当前几个重要的SNPs数据库,如dbSNP、1000Genome、genomAD、EXAC以及近期发表的中国人的SNPs数据库ChinaMap等,绝大多数只给出了人群AF指标,初步探索只有1KGP给出了2504个人的位点开源详细的基因型数据,可用于直接计算每个位点的真实人群纯合/杂合度。因此本次使用1KGP(phase3)进行计算差异探索。
为进一步考虑不同地区人种之间的差异,本次选择五类东亚人种(504个人的基因型)进行分析,包括CDX-西双版纳、CHB-北京汉族、CHS-南方汉族、JPT-日本东京、KHV-越南5个地区。
计算
1、直接计算杂合度
1KGP vcf文件中标注出每个SNPs位点上,每个人的基因型,用0、1进行判断纯杂合:ref纯合(0/0)、Alt纯合(1/1)、杂合(0/1、1/0)
target.vcf:
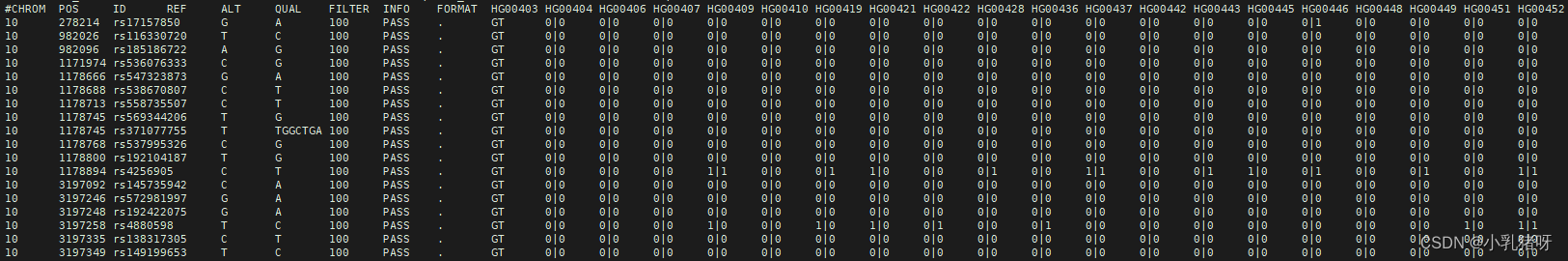
直接使用awk简单统计每个位点纯杂合,也可以用plink统计,但需要注意major、minor区分:
awk简单统计:
grep -v ^“#” target.vcf | awk -v OFS=“\t” ‘{ count_hot1 = gsub(/0|0/, “”); count_het = gsub(/1|0|0|1/, “”);count_hot2 = gsub(/1|1/, “”); print $1,$2,$2+1,$3,$4,$5,count_hot1,count_het,count_hot2}’ | awk ‘{if(length($5)<2 && length($6)<2 ) print $0 }’ > freq.bed
plink统计(结果感觉有点问题,还不如自己写个小脚本):
vcftools --vcf target.vcf --plink --out target.recode
plink --file target.recode --hardy
freq.bed:最后三列分别为纯杂合的人群数
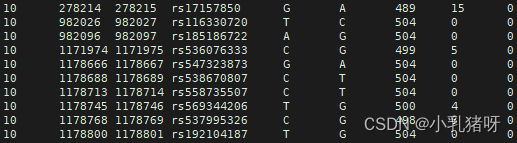
而后直接相除计算纯合、杂合度。
2、AF间接推导
杂合度=2AFx(1-AF)
相关性分析可以看到二者几乎无相关性:
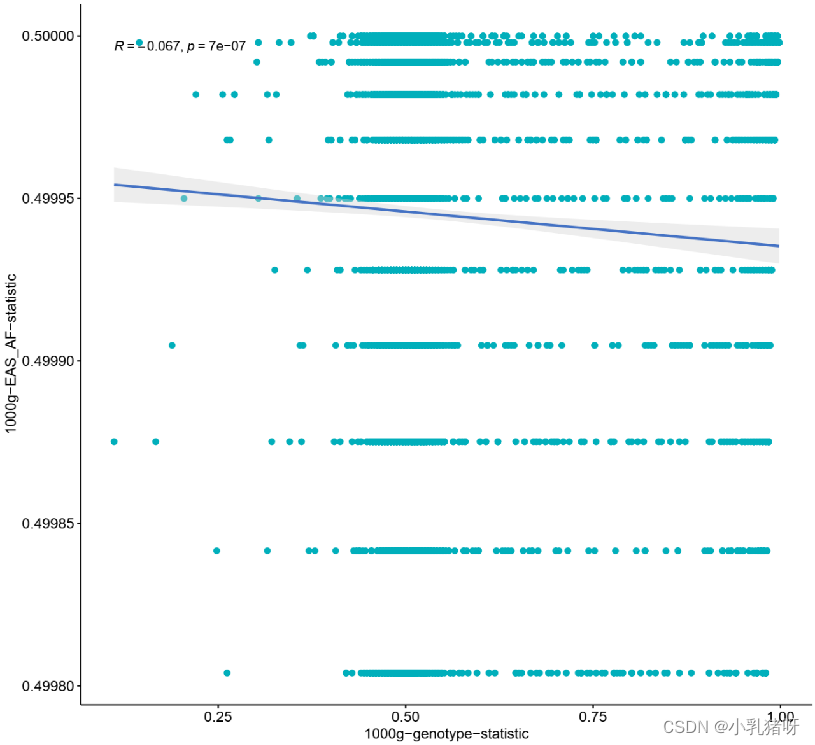
表明利用AF间接推导人群杂合度与实际差异较大。
不考虑遗传稳态,举一个极限的例子,一个位点所有人均为Aa,另一个位点AA、aa人群比例各占一半时,使用AF推导时,杂合度是一致的。即AF主要反映的是alt碱基比例,无法反应杂合人群概率。
结论:
考虑到位点人群差异、使用AF间接推导人群杂合度的方法不可取,一个较好的方法是借助千人基因组数据进行直接统计,如果1KGP不包括靶点SNPs,比较繁琐点的办法是找全基因组测序数据库,自己积累大样本队列进行统计















 5011
5011











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









