






#自留存档,有误请指出,感谢!!!
1. 首先要科学上网。
2. 进入Alphafold3官网AlphaFold Server (google.com),这里需要进行账号注册,需要谷歌邮箱账号,只要科学上网后就可以申请账号。注:目前国内手机号可以用,所以注册过程很快。
3. 确定蛋白序列:我这里是从Uniport上下载的蛋白序列。
4. 导入数据进行分析,数据类型也可以是RNA或者DNA序列。分析的时间和你导入的序列大小相关,我尝试过转录因子和启动子的预测,由于序列较小,整个过程只需要不到10分钟。
5. 分析结果,强烈建议先去查看结果说明
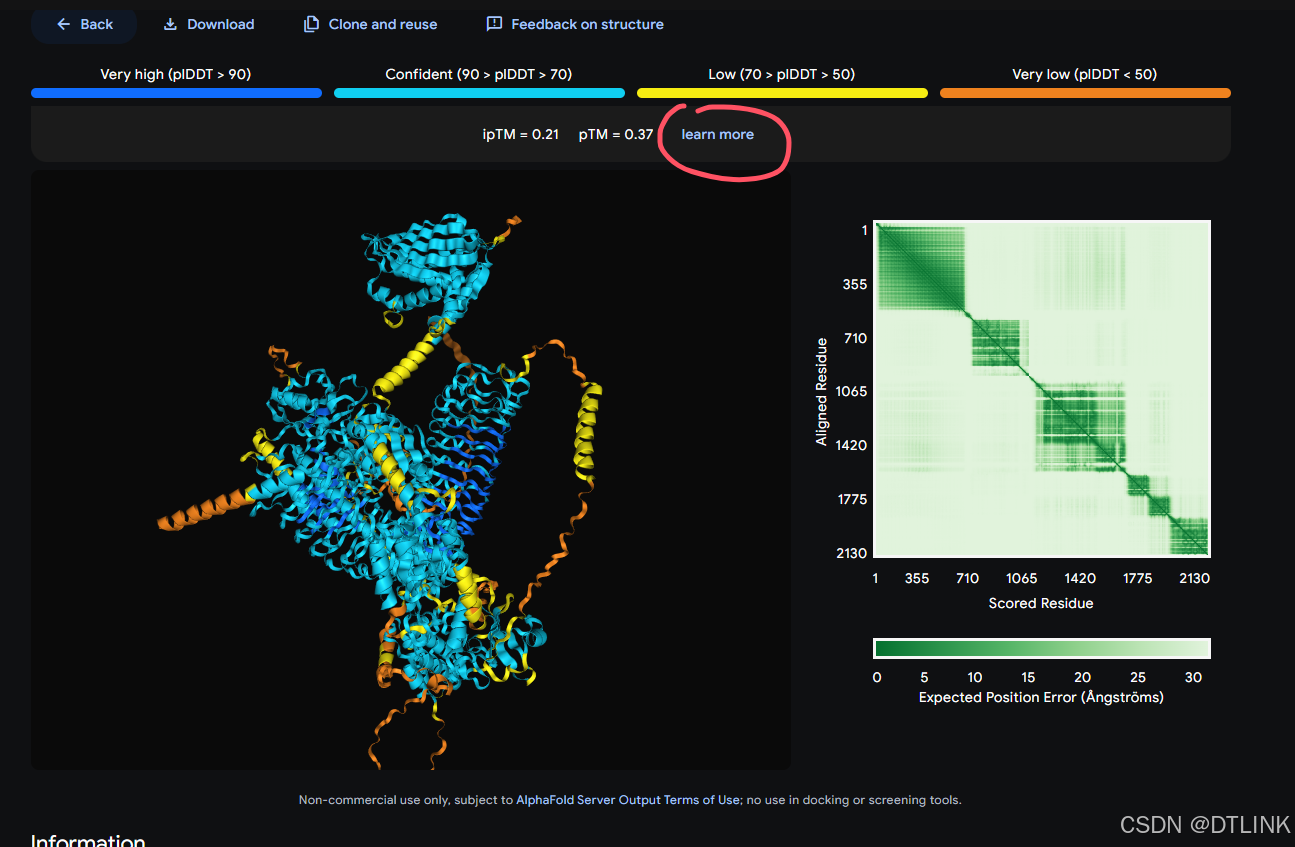
6. 下载互作结果数据,用于后续分析
7. 下载的数据里面,我们把cif文件用pymol打开并查看互作位点
8. pymol可以从官网下载PyMOL | pymol.org
9. 经过修改处理后可以清晰看到预测互作位点,这时就可以结合文献资料进行互作结果的解读

------------------------------------------------------------------------------------------------------------------
2024年11月27日更新
接着上面的内容,添加具体如何使用pymol来查看蛋白的互作位点
下载好的互作结果文件中有多个cif文件,这是后续要使用的文件,model后面有数字,这是预测的多个结果,我看网上的教程说数字越小代表可信度越高,但是也不绝对,各位如果有可靠的已经发表或者自己的生化实验结果可以对照查看,这里我就使用model_0来作为示例来进行。
1. 打开pymol软件并打开cif文件。或者把cif文件直接拖拽到pymol软件图标上。打开后可以直接看到蛋白的结构图。pymol的操作按理说有很多教程,这里我也是直接把别人的视频教程整理成了文字档。
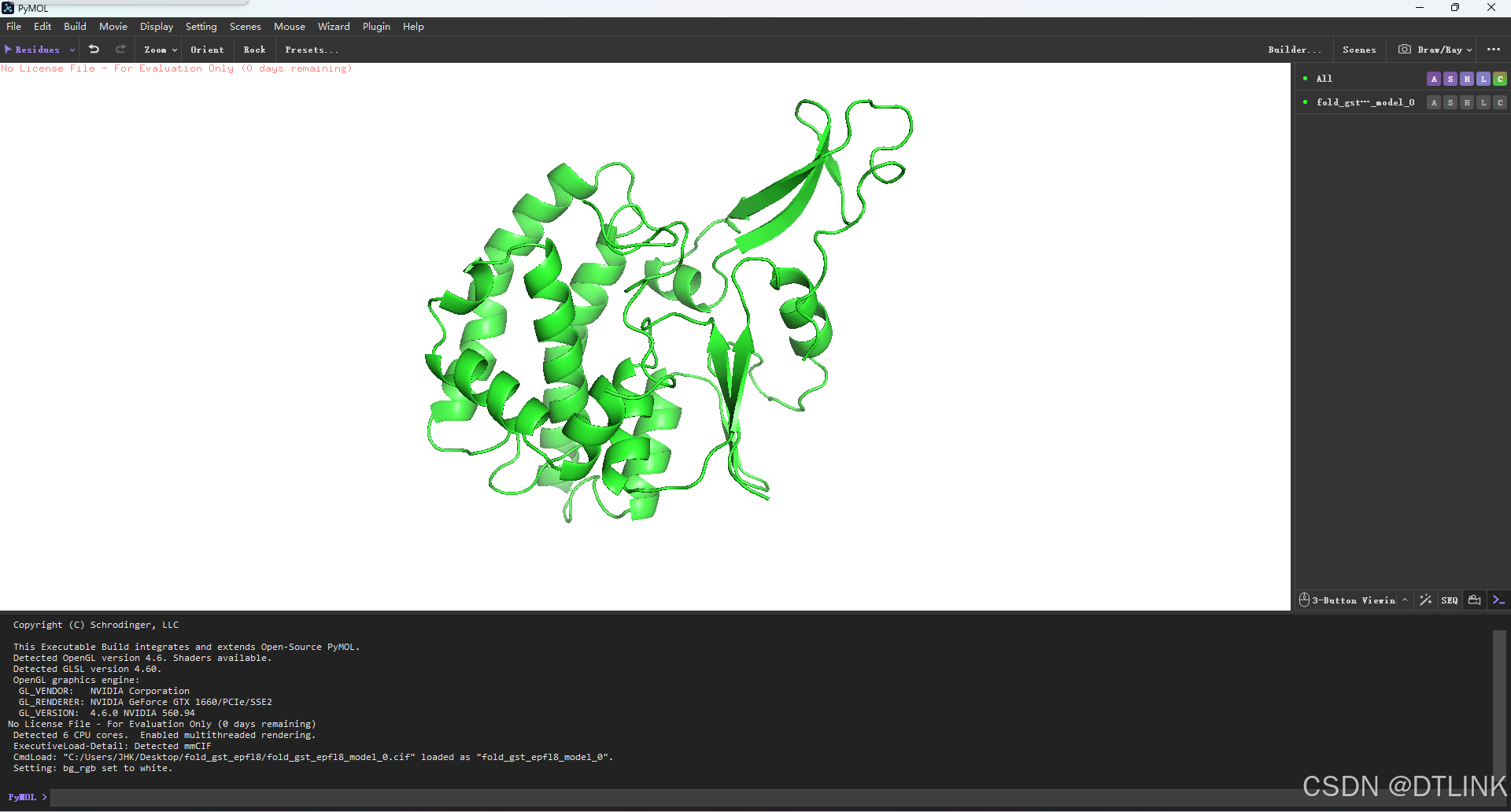
2. 更改背景色
Display——Background——white
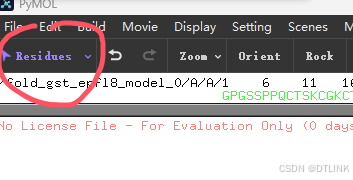
3. 更改蛋白对象名称和改变目标蛋白颜色
首先在这里点击将选项改为Chains,这样改变后点击蛋白可以选择整个肽链。
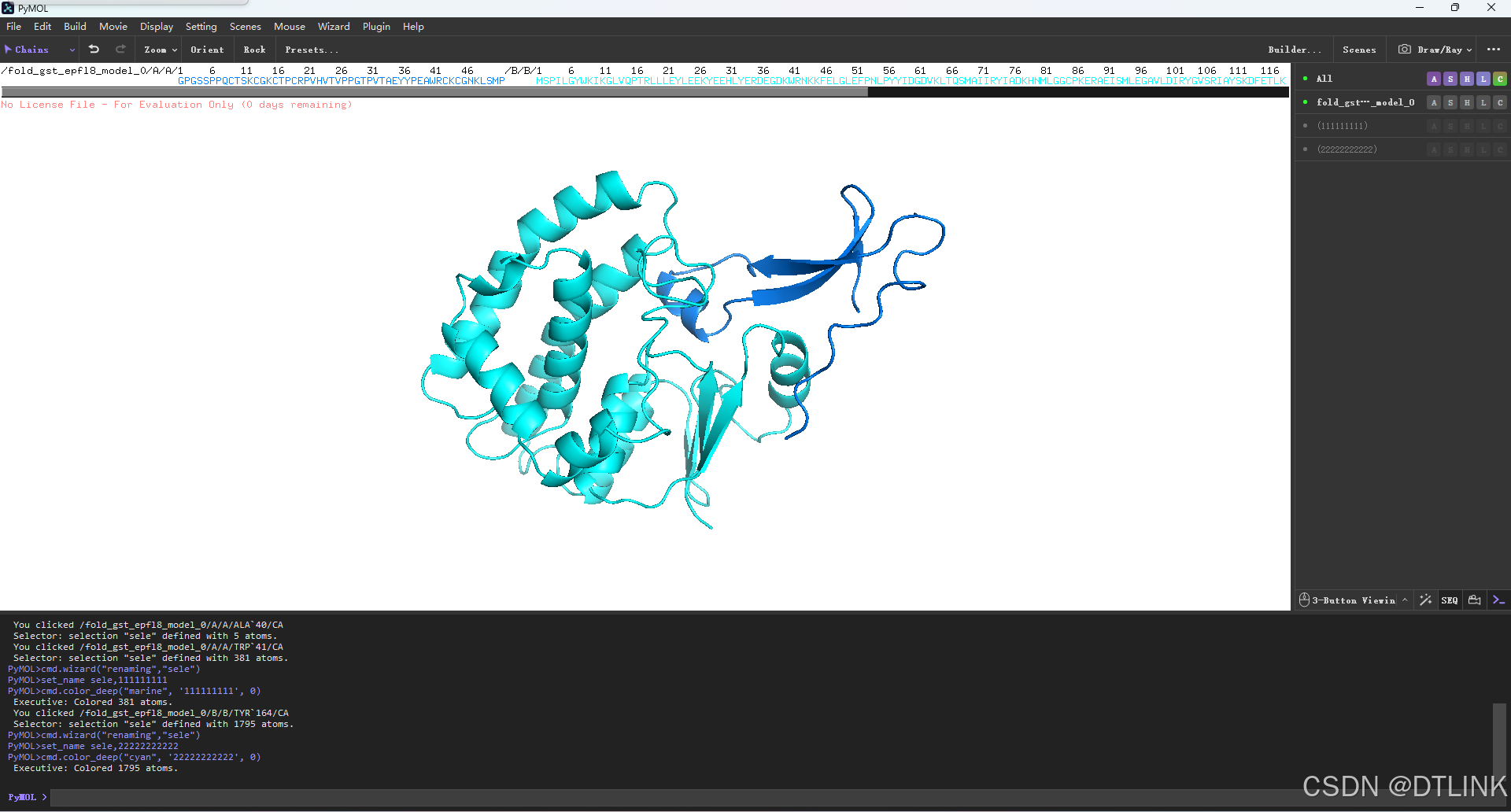
选择好之后,右侧的工作框会出现新的项目,点击该项目右侧的A图标来进行rename更名。点击C图标进行改色,这个颜色怎么改就看各自需求。需要注意的是在对一个蛋白进行编辑时,如果是选中状态是明显高亮的,没有选中则是灰色。后续在对不同蛋白进行编辑时需要注意这点,不然编辑对象可能出错。
这里我使用的是海军蓝和松石绿。
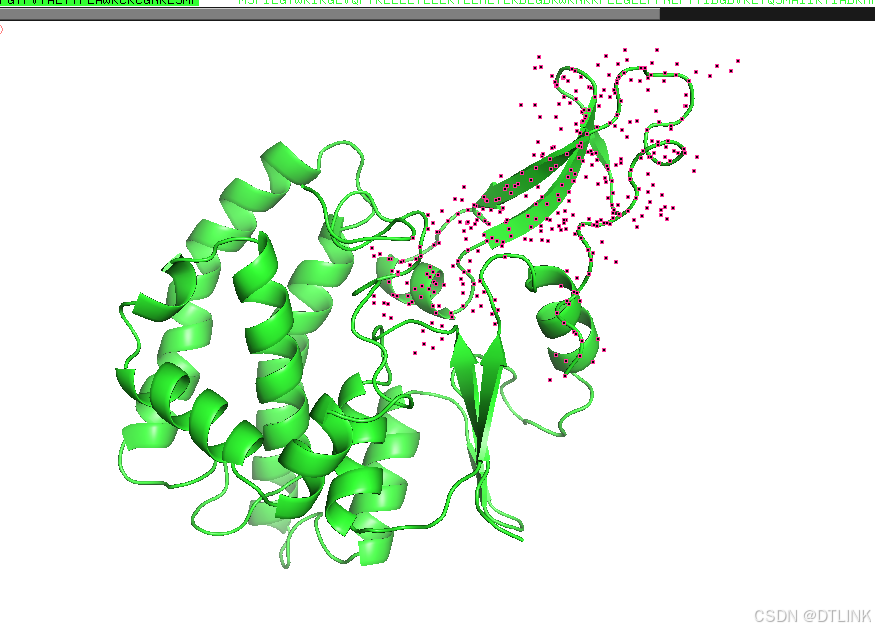
4. 查看蛋白互作位点
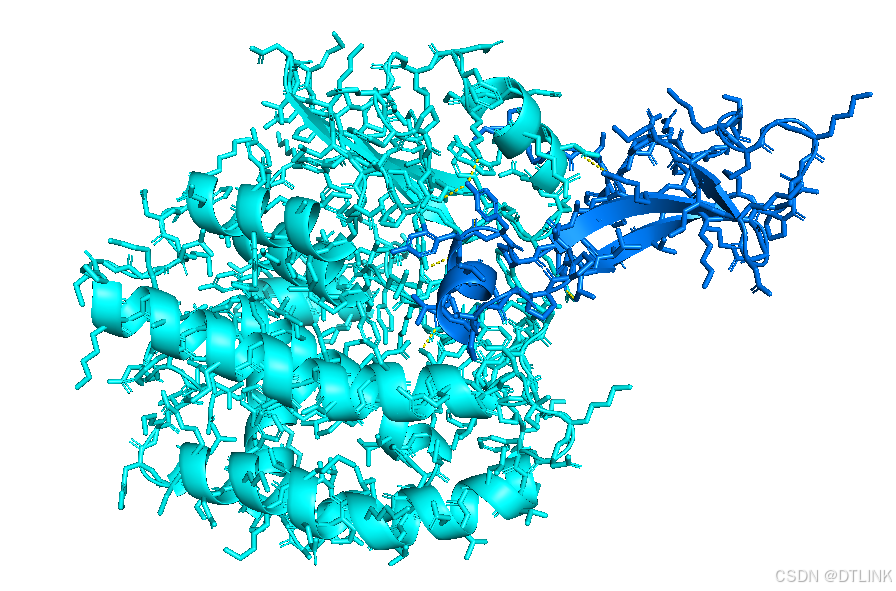
随便选中一条肽链进行编辑。步骤是A——find——polar contacts——to other atoms in object
之后就会出现类似这种黄色的小棒
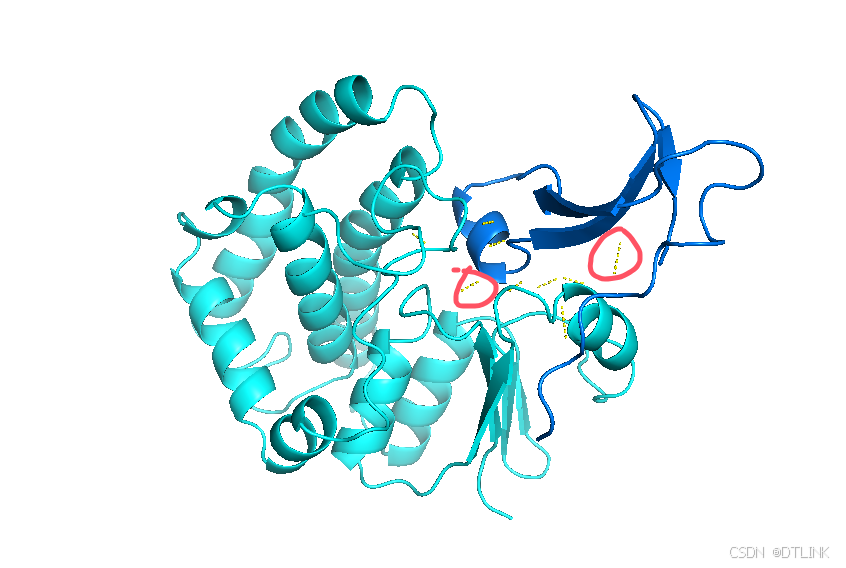
先把每条肽链的表示方式换成棍状模式,步骤是S——sticks
5. 对氨基酸残基进行编辑
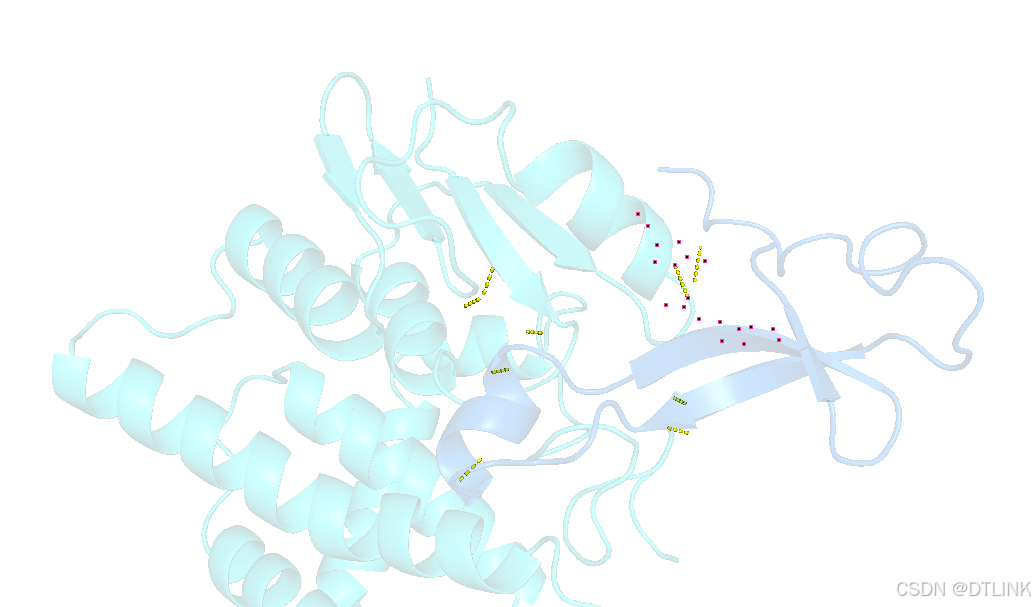
同样的操作,将左上角的chains改为Residues,这样我们就可以对氨基酸残基进行选中。最好是把所有的互作都选中,我这里偷个懒,只选了一个。
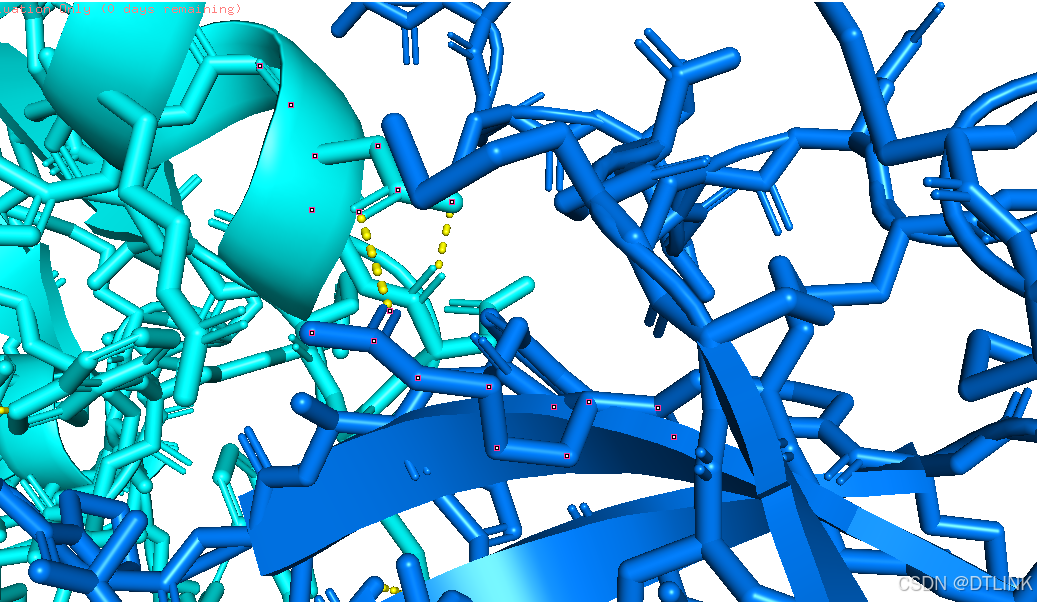
之后隐藏棍状,步骤还是在右侧项目栏中操作。H——sticks

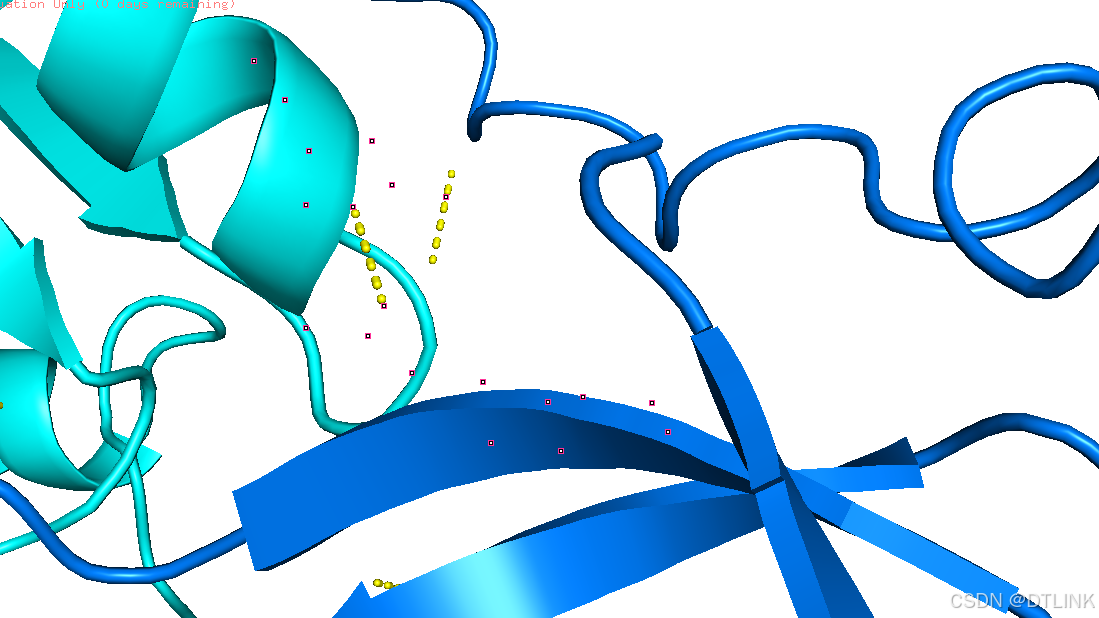
为了凸显出这个互作位点,我们再把主体蛋白模型进行透明化。步骤:setting——Transparence——cartoon——80%
选中残基图层再将其棍状结构显示出来。
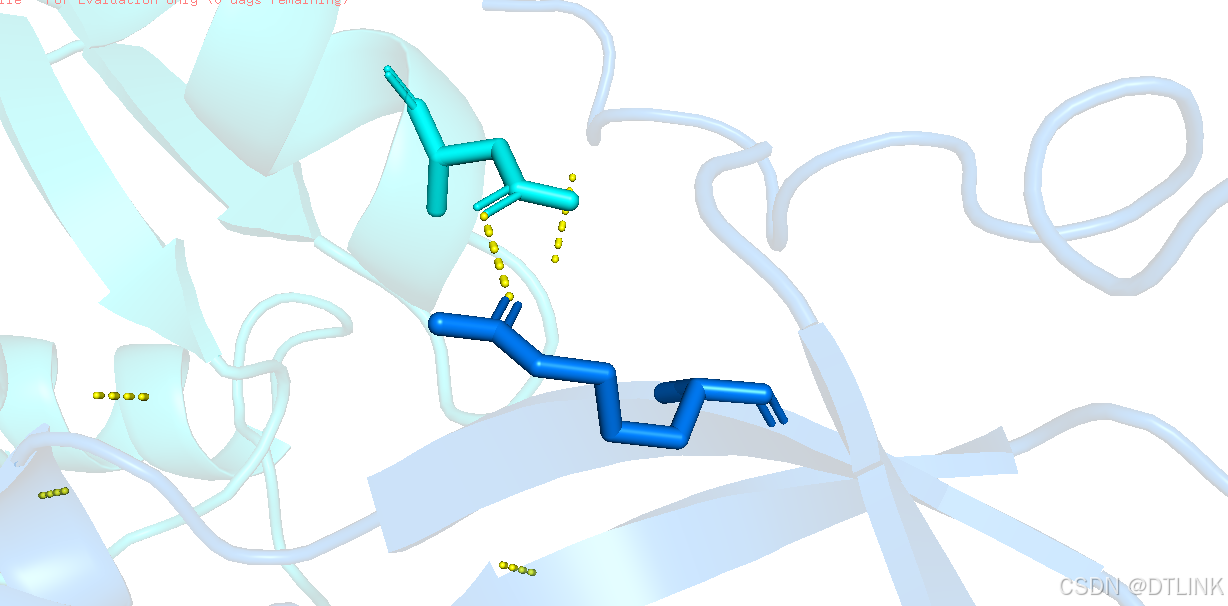
之后再进行编辑将其互作位点的氨基酸信息标注出来,步骤:L——residues
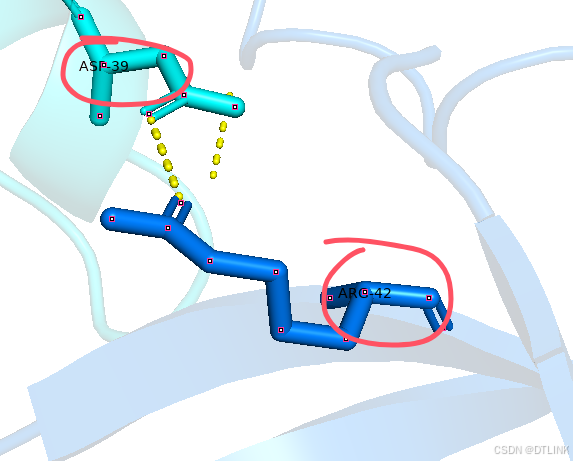
当位点很多时,这样不好看,可以再将这些氨基酸信息进行编辑,切换到编辑模式button editing,再按住ctrl用鼠标推拽就可以了。

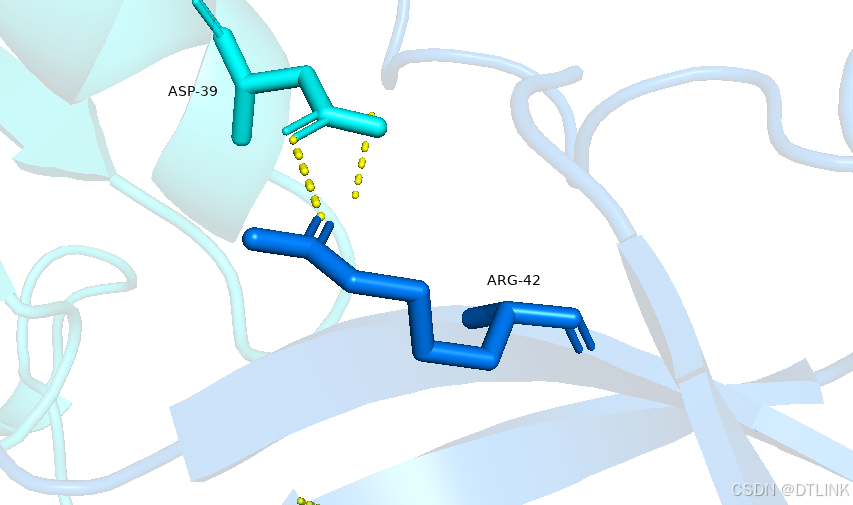
再将氨基酸信息与对应位点进行连接,setting——label——show connector,这样就更直观了。
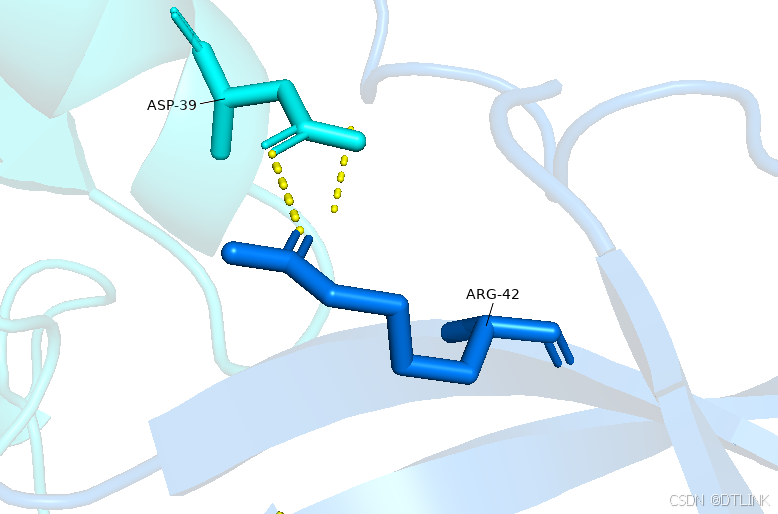
怎么去看具体的序列信息???也就是互作位点是哪个氨基酸?
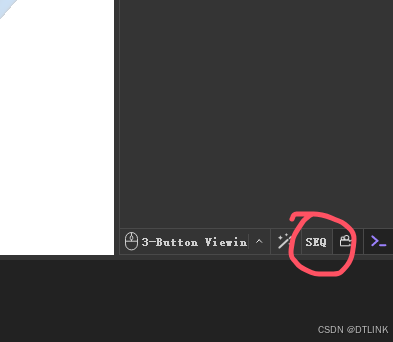
1. 右下角有个seq选项。点击之后模型上方就会出现序列框。
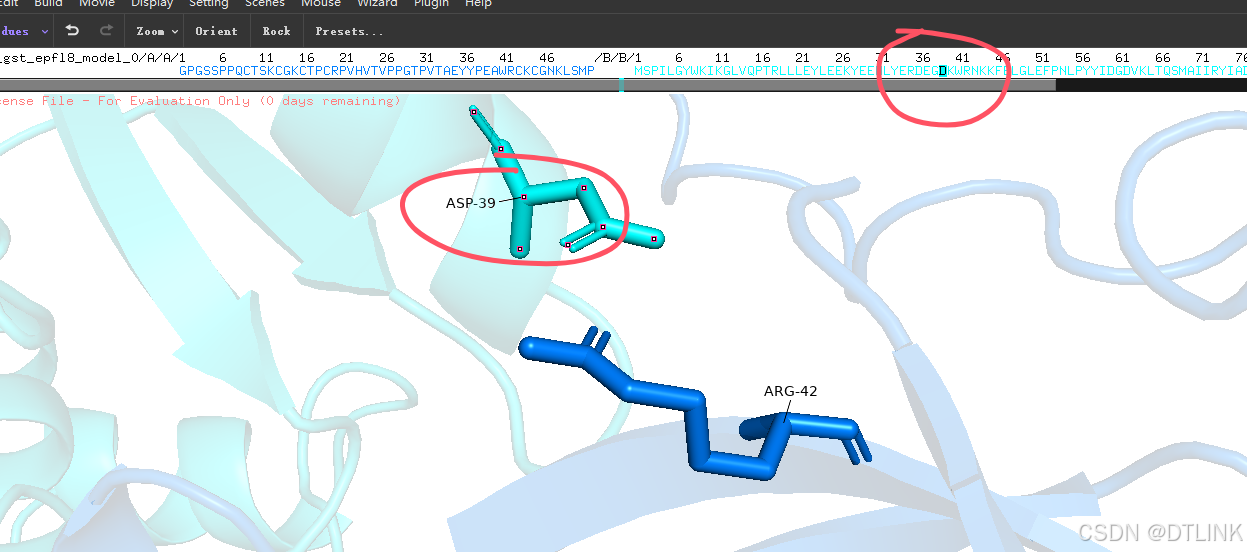
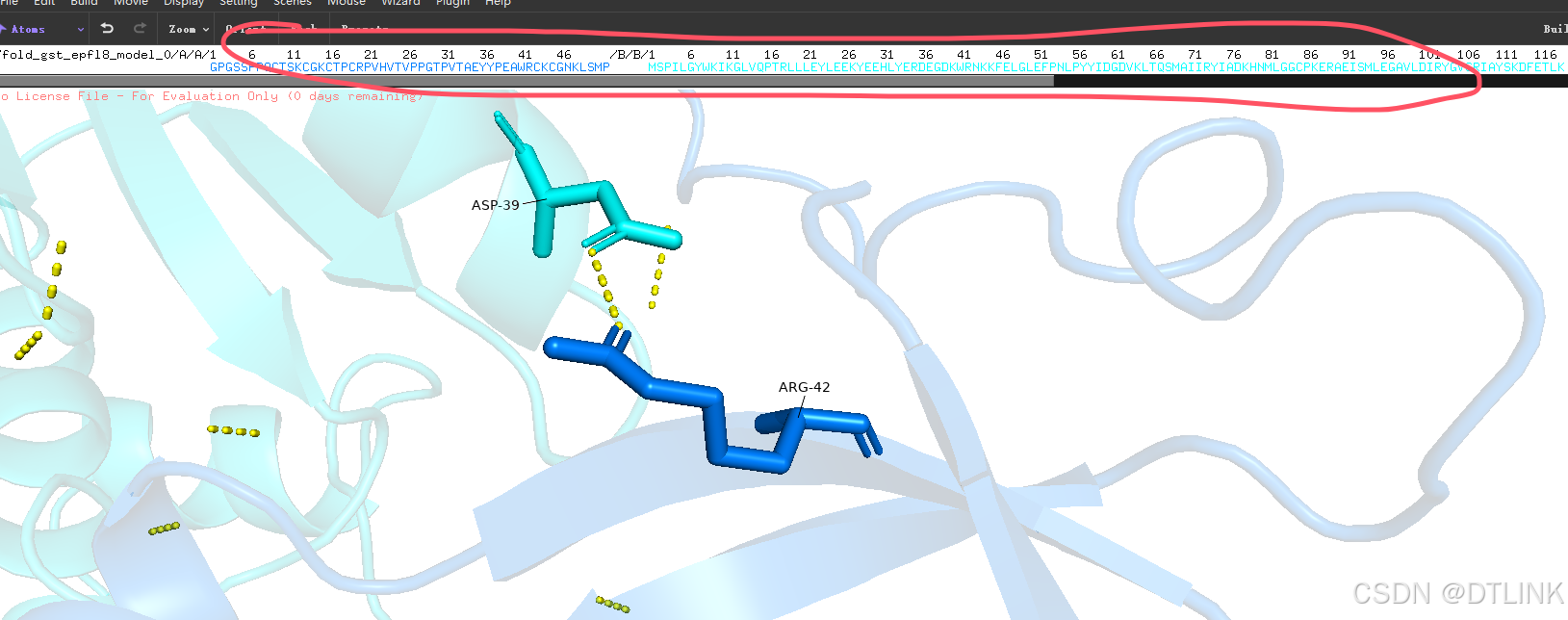 2. 再次把鼠标改为view模式,点击这个互作位点,就可以具体显示在序列上的哪个位置,当然这里其实也已经显示了,比如说ASP-39就是说明这条序列上的第39位天冬氨酸
2. 再次把鼠标改为view模式,点击这个互作位点,就可以具体显示在序列上的哪个位置,当然这里其实也已经显示了,比如说ASP-39就是说明这条序列上的第39位天冬氨酸

















 1918
1918

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









