






研究蛋白功能的时候,常常会遇到一个点突就会导致蛋白功能的变化这种情况,除了使用常规的生化或遗传方式去研究以外,我们还可以用蛋白构象变化来进行补充说明。这里我分析了两个单个氨基酸改变的蛋白之间的构象变化。如有不足,请各位补充~~~
--------------------------------------------------------------------------------------------------------------------------
这一步不进行图片展示,可参考此前文章。
本次操作是使用开源版pymol,其实看了一下开源的版本和不开源版本都挺好用,至少都满足我现阶段的使用。
1. 自备两种蛋白的氨基酸序列。
2. 科学上网,进入alphfold3进行蛋白结构预测,并下载预测结果。
3. 在pymol中打开cif文件,并选择可信度最高的文件(model_0)进行后续分析。
---------------------------------------------------------------------------------------------------------------------------
pymol中的具体操作过程:
1. 打开protein_A和peotein_Amut文件,这时两个蛋白会存在一个界面中
2. 进行两个蛋白序列的比对,在命令行输入:align protein_A, protein_Amut
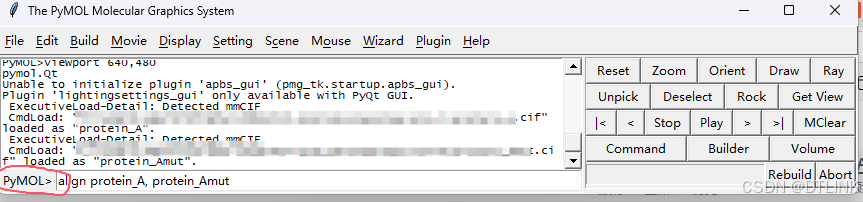
3. 初步观察结构,发现A和Amut其实在空间位置上已经不能实现高度重合,那如何实现量化展示?
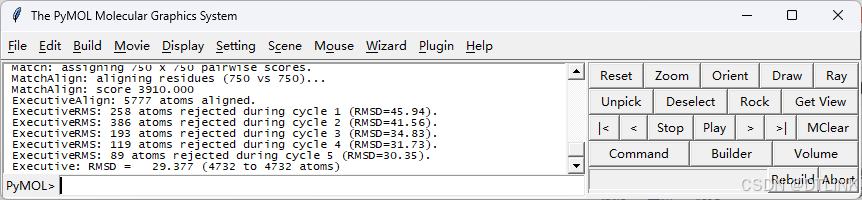
4. 量化的结果其实已经展示在命令行上方界面,这里的RMSD(均方根偏差,Root Mean Square Deviation)就已经说明二者之间存在区别。RMSD值越小,表示两个蛋白质结构越相似;反之,RMSD值越大,表示它们之间的差异越显著。
这里直接复制AI给的答案,各位自行衡量~最好还是引用一些相关文献作为支持!
AI:RMSD值的显著性阈值并没有绝对的标准,因为它取决于具体的研究背景和所比较的蛋白质类型。然而,一般来说,以下几个范围可以作为大致的参考:
- **RMSD < 1.0 Å**:通常被认为是非常相似的结构,结构差异较小。
- **1.0 Å ≤ RMSD < 2.0 Å**:表示相似但有一定的结构差异,可以认为这些蛋白质在整体上仍保持相似的构象。
- **2.0 Å ≤ RMSD < 4.0 Å**:通常被视为显著差异的结构,可能表明蛋白质的折叠或功能上存在明显变化。
- **RMSD ≥ 4.0 Å**:通常表示蛋白质结构之间存在显著差异,可能反映出不同的功能或生物学角色。
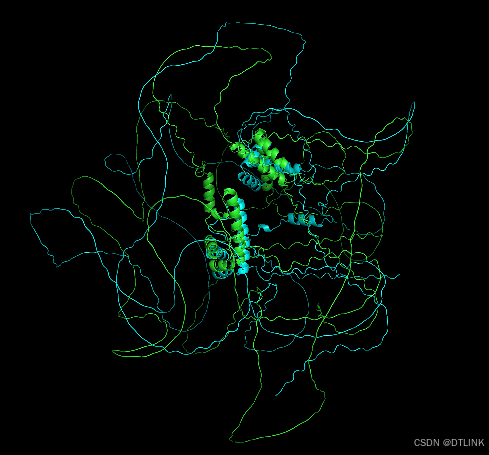
5. 最后,我们在把对应突变位点进行美化突出显示就完成了,这里的操作步骤就不进行展示了,可参考前面的文章。


















 8021
8021

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









