







 PeSTo是一个无参数的几何Transformer,用于蛋白质结构分析,尤其在预测蛋白质与其他分子的结合界面方面表现出高精度和效率。模型直接作用于原子坐标,无需物理化学属性,能处理大量结构数据,包括MD模拟和折叠体预测,为揭示蛋白质相互作用的动态特性提供了新途径。
PeSTo是一个无参数的几何Transformer,用于蛋白质结构分析,尤其在预测蛋白质与其他分子的结合界面方面表现出高精度和效率。模型直接作用于原子坐标,无需物理化学属性,能处理大量结构数据,包括MD模拟和折叠体预测,为揭示蛋白质相互作用的动态特性提供了新途径。
Title:PeSTo: parameter-free geometric deep learning for accurate prediction of protein binding interfaces
期刊:nature communication
分区:一区
影响因子:16.6
webserver:t Pesto
Github:GitHub - LBM-EPFL/PeSTo
摘要
蛋白质是生命的重要分子组成部分,由于其特定的分子相互作用,负责大多数生物功能。然而,预测它们的绑定接口仍然是一个挑战。在这项研究中,我们提出了一个几何变换器,它直接作用于仅标有元素名称的原子坐标。由此产生的模型——蛋白质结构转换器,PeSTo——在预测蛋白质-蛋白质界面方面超越了当前的技术水平,并且还可以高度自信地预测和区分涉及核酸、脂质、离子和小分子的界面。它的低计算成本使得能够处理大量的结构数据,例如分子动力学系综,允许发现在静态实验解决的结构中保持不明显的界面。此外,由从头结构预测提供的不断增长的折叠体可以很容易地被分析,为揭示未探索的生物学提供了新的机会。
方法和数据集
数据集
数据集由来自蛋白质数据库的所有生物组件组成。使用簇之间最大30%的序列同一性来聚集亚基。亚基簇被分成大约70%的训练集(376216条链),15%的验证集(101700条链),和15%的测试集(97424条链)。我们通过评估验证集上的模型来选择最佳超参数。测试集由包含来自MaSIF-site基准数据集的53个亚单位或来自蛋白质-蛋白质对接基准5.038 (PPDB5)数据集的230个结构中的任何一个的簇组成。此外,我们提取了在ScanNet15的基准数据集和PeSTo的测试数据集中常见的子集417结构。除非特别说明,所有被选择用来评估模型预测质量的例子都属于测试集。
特征和标签
我们确定了PDB上30种最常见的原子元素。元素被用作onehot编码。输入矢量要素最初设置为零。距离矩阵和归一化位移向量矩阵被用作几何特征。氨基酸、核酸、离子、配体和脂质分别选自20、8、16、31和4种最常见的分子。用于帮助解决结构的非天然分子被忽略。界面被定义为5英寸以内的残基-残基接触。所有蛋白质-蛋白质界面以及蛋白质-核酸、蛋白质-离子、蛋白质-配体和蛋白质-脂质界面都被识别。每个子单元的接口细节作为交互类型矩阵(79×79)存储在数据集中。这使得能够在训练会话开始时选择特定接口作为标签,而不必重建整个数据集。界面靶标可以从79个可用分子的子集的任意组合中选择。
方法
蛋白质结构的Transformer架构
输入特征被嵌入到一个输入状态大小为S = 32的三层神经网络中,其中隐层大小为32。每个几何变换器由3层的5个神经网络组成,以执行补充算法1中描述的多头自关注(S = 32,Nkey = 3,Nhead = 2)。对于原子数量小于设置的最近邻(nn)数量的结构,额外的不存在的相互作用被发送到具有设
置为零的标量和矢量状态的汇聚节点。连续应用4组8个几何变换器,每组的最近邻数量递增(nn = 8、16、32和64)。如补充算法2中所述,通过在形成每个残基(S = 32,Nhead = 4)的原子上使用局部多头掩码,几何残基汇集模块将结构的原子级编码聚集成残基级描述。最后一个模块是一个多层感知器,具有3层S = 32的隐藏大小,解码所有残差的状态并计算预测,返回从0到1的置信度得分。
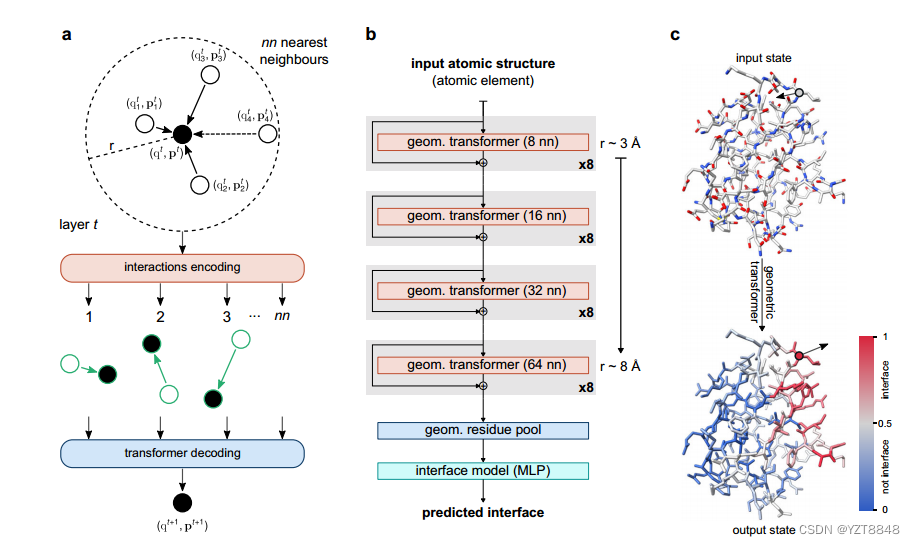
Transformer的蛋白质结构(PesTo)
许多成功的方法结合了transformers和几何深度学习,将结构表示为图形或点云,并整合了神经网络的不变性或等方差要求。主要突破来自蛋白质折叠领域,其中AlphaFold将注意力整合到Evoformer模块和结构模块中,RoseTTAFold20模型的第三个轨道使用阿瑟(3)转换器在折叠过程中细化原子坐标。此外,递归几何网络 (RGN2)利用Frenet-Serret公式来表示蛋白质的主链,几何矢量感知器 (GVP)使用线性运算来构成带有gating的矢量特征。已经开发了多种其他基于机器学习的蛋白质-蛋白质相互作用位点预测方法。
我们在这里介绍PeSTo,一个无参数的几何变换器,直接作用于蛋白质结构的原子。如图1所示以及在方法中详细描述的,该结构被表示为以原子位置为中心的点云,
 最低0.47元/天 解锁文章
最低0.47元/天 解锁文章


















 1万+
1万+

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









