






 本文详细介绍qiime2R包的使用方法,包括如何将qiime2中的特征表、分类文件及进化树文件导入R软件,进行图形绘制及统计分析。文章通过Movingpictures教程数据,演示了读取qza文件、构建Phyloseq对象、计算α多样性、绘制PCoA图、制作热图和柱状图等操作,并展示了如何进行差异性丰度计算和绘制造型树。
本文详细介绍qiime2R包的使用方法,包括如何将qiime2中的特征表、分类文件及进化树文件导入R软件,进行图形绘制及统计分析。文章通过Movingpictures教程数据,演示了读取qza文件、构建Phyloseq对象、计算α多样性、绘制PCoA图、制作热图和柱状图等操作,并展示了如何进行差异性丰度计算和绘制造型树。
qiime2R包讲qiime2中的最终特征表文件(table.qza),分类文件(taxonomy.qza)及进化树文件直接导入R软件中,进行图形的绘制及统计分析。
功能如下:
read_qza()- Function for reading artifacts (.qza).qza_to_phyloseq()- Imports multiple artifacts to produce a phyloseq object.read_q2metadata()- Reads qiime2 metadata file (containing q2-types definition line)write_q2manifest()- Writes a read manifest file to import data into qiime2theme_q2r()- A ggplot2 theme for publication-type figures.print_provenance()- A function to display provenance information.is_q2metadata()- A function to check if a file is a qiime2 metadata file.parse_taxonomy()- A function to parse taxonomy strings and return a table where each column is a taxonomic class.parse_ordination()- A function to parse the internal ordination format.read_q2biom- A function for reading QIIME2 biom files in format v2.1make_clr- Transform feature table using centered log2 ratio.make_proportion- Transform feature table to proportion (sum to 1).make_percent- Transform feature to percent (sum to 100).interactive_table- Create an interactive table in Rstudio viewer or rmarkdown html.summarize_taxa- Create a list of tables with abundances sumed to each taxonomic level.taxa_barplot- Create a stacked barplot using ggplot2.taxa_heatmap- Create a heatmap of taxonomic abundances using gplot2
qiime2R包的安装
if (!requireNamespace("devtools", quietly = TRUE)){install.packages("devtools")}
devtools::install_github("jbisanz/qiime2R")读取qza文件(这里以Moving pictures tutorial数据为例)
https://docs.qiime2.org/2020.8/tutorials/moving-pictures/
SVs<-read_qza("table.qza")
names(SVs)
[1] "uuid" "type" "format" "contents" "version"
[6] "data" "provenance"
SVs$data[1:5,1:5] #show first 5 samples and first 5 taxa
SVs$uuid
[1] "706b6bce-8f19-4ae9-b8f5-21b14a814a1b"
SVs$type
[1] "FeatureTable[Frequency]"读取metadata
metadata<-read_q2metadata("sample-metadata.tsv")
head(metadata) # show top lines of metadata
# SampleID barcode-sequence body-site year month day subject reported-antibiotic-usage days-since-experiment-start
#2 L1S8 AGCTGACTAGTC gut 2008 10 28 subject-1 Yes 0
#3 L1S57 ACACACTATGGC gut 2009 1 20 subject-1 No 84
#4 L1S76 ACTACGTGTGGT gut 2009 2 17 subject-1 No 112
#5 L1S105 AGTGCGATGCGT gut 2009 3 17 subject-1 No 140
#6 L2S155 ACGATGCGACCA left palm 2009 1 20 subject-1 No 84
#7 L2S175 AGCTATCCACGA left palm 2009 2 17 subject-1 读取taxonomy
taxonomy<-read_qza("taxonomy.qza")
head(taxonomy$data)
# Feature.ID Taxon Confidence
#1 4b5eeb300368260019c1fbc7a3c718fc k__Bacteria; p__Bacteroidetes; c__Bacteroidia; o__Bacteroidales; f__Bacteroidaceae; g__Bacteroides; s__ 0.9972511
#2 fe30ff0f71a38a39cf1717ec2be3a2fc k__Bacteria; p__Proteobacteria; c__Betaproteobacteria; o__Neisseriales; f__Neisseriaceae; g__Neisseria 0.9799427
#3 d29fe3c70564fc0f69f2c03e0d1e5561 k__Bacteria; p__Firmicutes; c__Bacilli; o__Lactobacillales; f__Streptococcaceae; g__Streptococcus 1.0000000
#4 868528ca947bc57b69ffdf83e6b73bae k__Bacteria; p__Bacteroidetes; c__Bacteroidia; o__Bacteroidales; f__Bacteroidaceae; g__Bacteroides; s__ 0.9955859
#5 154709e160e8cada6bfb21115acc80f5 k__Bacteria; p__Bacteroidetes; c__Bacteroidia; o__Bacteroidales; f__Bacteroidaceae; g__Bacteroides 1.0000000
#6 1d2e5f3444ca750c85302ceee2473331 k__Bacteria; p__Proteobacteria; c__Gammaproteobacteria; o__Pasteurellales; f__Pasteurellaceae; g__Haemophilus; s__parainfluenzae 0.9455365
taxonomy<-parse_taxonomy(taxonomy$data)
head(taxonomy)
# Kingdom Phylum Class Order Family Genus Species
#4b5eeb300368260019c1fbc7a3c718fc Bacteria Bacteroidetes Bacteroidia Bacteroidales Bacteroidaceae Bacteroides <NA>
#fe30ff0f71a38a39cf1717ec2be3a2fc Bacteria Proteobacteria Betaproteobacteria Neisseriales Neisseriaceae Neisseria <NA>
#d29fe3c70564fc0f69f2c03e0d1e5561 Bacteria Firmicutes Bacilli Lactobacillales Streptococcaceae Streptococcus <NA>
#868528ca947bc57b69ffdf83e6b73bae Bacteria Bacteroidetes Bacteroidia Bacteroidales Bacteroidaceae Bacteroides <NA>
#154709e160e8cada6bfb21115acc80f5 Bacteria Bacteroidetes Bacteroidia Bacteroidales Bacteroidaceae Bacteroides <NA>
#1d2e5f3444ca750c85302ceee2473331 Bacteria Proteobacteria Gammaproteobacteria Pasteurellales Pasteurellaceae Haemophilus parainfluenzae
构建 Phyloseq
physeq<-qza_to_phyloseq(
features="inst/artifacts/2020.2_moving-pictures/table.qza",
tree="inst/artifacts/2020.2_moving-pictures/rooted-tree.qza",
taxonomy="inst/artifacts/2020.2_moving-pictures/taxonomy.qza",
metadata = "inst/artifacts/2020.2_moving-pictures/sample-metadata.tsv"
)
physeq
## phyloseq-class experiment-level object
## otu_table() OTU Table: [ 759 taxa and 34 samples ]
## sample_data() Sample Data: [ 34 samples by 10 sample variables ]
## tax_table() Taxonomy Table: [ 759 taxa by 7 taxonomic ranks ]
## phy_tree() Phylogenetic Tree: [ 759 tips and 757 internal nodes ]
结果展示
Alpha diversity
library(tidyverse)
library(qiime2R)
metadata<-read_q2metadata("sample-metadata.tsv")
shannon<-read_qza("shannon_vector.qza")
shannon<-shannon$data %>% rownames_to_column("SampleID") # this moves the sample names to a new column that matches the metadata and allows them to be merged
gplots::venn(list(metadata=metadata$SampleID, shannon=shannon$SampleID))
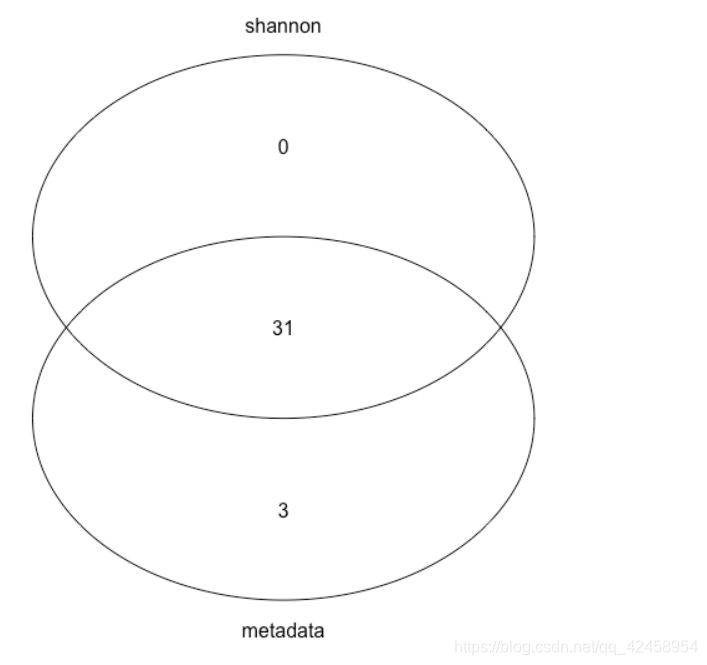
其中有3个样本无shannon值,接下来将进一步探索该三个样本
metadata<-
metadata %>%
left_join(shannon)
head(metadata)
# SampleID barcode-sequence body-site year month day subject reported-antibiotic-usage days-since-experiment-start shannon
#1 L1S8 AGCTGACTAGTC gut 2008 10 28 subject-1 Yes 0 3.188316
#2 L1S57 ACACACTATGGC gut 2009 1 20 subject-1 No 84 3.985702
#3 L1S76 ACTACGTGTGGT gut 2009 2 17 subject-1 No 112 3.461625
#4 L1S105 AGTGCGATGCGT gut 2009 3 17 subject-1 No 140 3.972339
#5 L2S155 ACGATGCGACCA left palm 2009 1 20 subject-1 No 84 5.064577
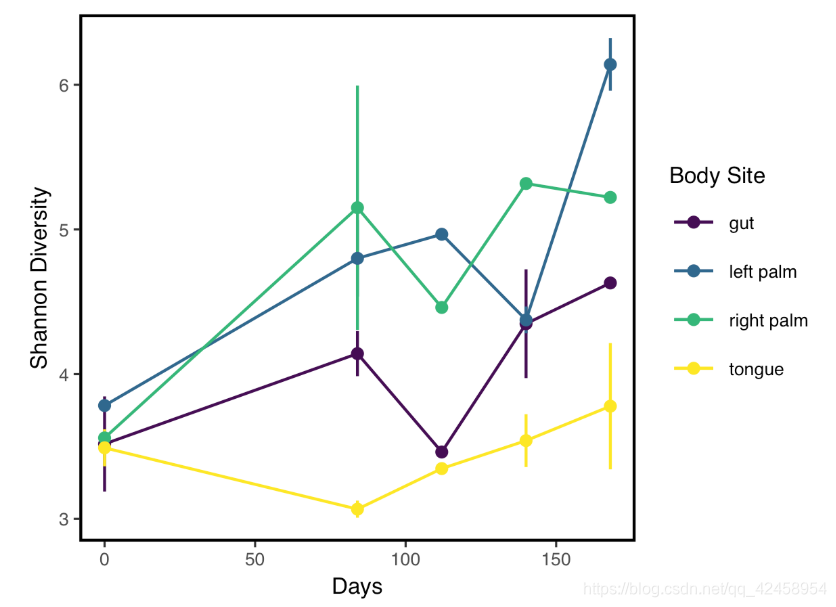
#6 L2S175 AGCTATCCACGA left palm 2009 2 17 subject-1 metadata %>%
filter(!is.na(shannon)) %>%
ggplot(aes(x=`days-since-experiment-start`, y=shannon, color=`body-site`)) +
stat_summary(geom="errorbar", fun.data=mean_se, width=0) +
stat_summary(geom="line", fun.data=mean_se) +
stat_summary(geom="point", fun.data=mean_se) +
xlab("Days") +
ylab("Shannon Diversity") +
theme_q2r() + # try other themes like theme_bw() or theme_classic()
scale_color_viridis_d(name="Body Site") # use different color scale which is color blind friendly
ggsave("Shannon_by_time.pdf", height=3, width=4, device="pdf") # save a PDF 3 inches by 4 inches
抗生素使用对多样性的影响
metadata %>%
filter(!is.na(shannon)) %>%
ggplot(aes(x=`reported-antibiotic-usage`, y=shannon, fill=`reported-antibiotic-usage`)) +
stat_summary(geom="bar", fun.data=mean_se, color="black") + #here black is the outline for the bars
geom_jitter(shape=21, width=0.2, height=0) +
coord_cartesian(ylim=c(2,7)) + # adjust y-axis
facet_grid(~`body-site`) + # create a panel for each body site
xlab("Antibiotic Usage") +
ylab("Shannon Diversity") +
theme_q2r() +
scale_fill_manual(values=c("cornflowerblue","indianred")) + #specify custom colors
theme(legend.position="none") #remove the legend as it isn't needed
ggsave("../../../images/Shannon_by_abx.pdf", height=3, width=4, device="pdf") # save a PDF 3 inches by 4 inches
其中两个样本间多样性的比较
metadata %>%
filter(!is.na(shannon)) %>%
ggplot(aes(x=subject, y=shannon, fill=`subject`)) +
stat_summary(geom="bar", fun.data=mean_se, color="black") + #here black is the outline for the bars
geom_jitter(shape=21, width=0.2, height=0) +
coord_cartesian(ylim=c(2,7)) + # adjust y-axis
facet_grid(~`body-site`) + # create a panel for each body site
xlab("Antibiotic Usage") +
ylab("Shannon Diversity") +
theme_q2r() +
scale_fill_manual(values=c("cornflowerblue","indianred")) + #specify custom colors
theme(legend.position="none") #remove the legend as it isn't needed
ggsave("Shannon_by_person.pdf", height=3, width=4, device="pdf") # save a PDF 3 inches by 4 inches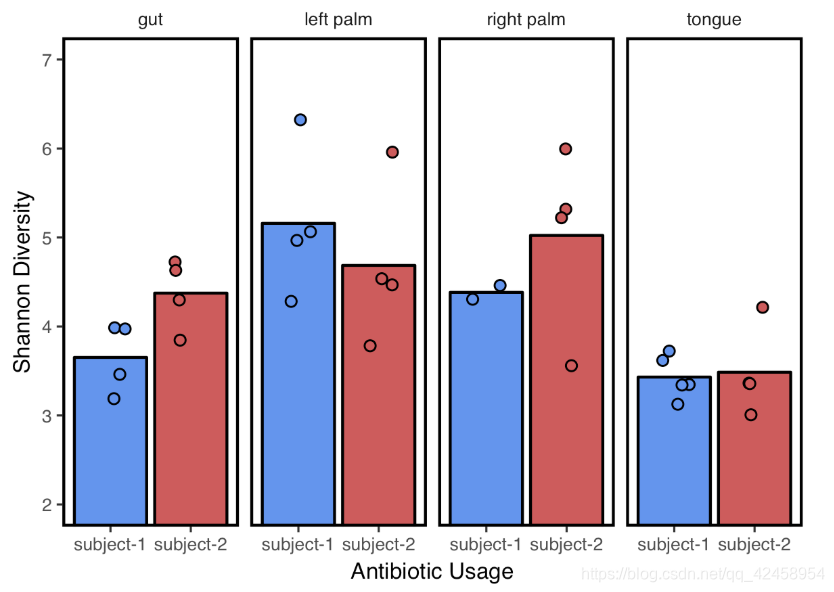
PCoA图的绘制
library(tidyverse)
library(qiime2R)
metadata<-read_q2metadata("sample-metadata.tsv")
uwunifrac<-read_qza("unweighted_unifrac_pcoa_results.qza")
shannon<-read_qza("shannon_vector.qza")$data %>% rownames_to_column("SampleID")
uwunifrac$data$Vectors %>%
select(SampleID, PC1, PC2) %>%
left_join(metadata) %>%
left_join(shannon) %>%
ggplot(aes(x=PC1, y=PC2, color=`body-site`, shape=`reported-antibiotic-usage`, size=shannon)) +
geom_point(alpha=0.5) + #alpha controls transparency and helps when points are overlapping
theme_q2r() +
scale_shape_manual(values=c(16,1), name="Antibiotic Usage") + #see http://www.sthda.com/sthda/RDoc/figure/graphs/r-plot-pch-symbols-points-in-r.png for numeric shape codes
scale_size_continuous(name="Shannon Diversity") +
scale_color_discrete(name="Body Site")
ggsave("PCoA.pdf", height=4, width=5, device="pdf") # save a PDF 3 inches by 4 inches 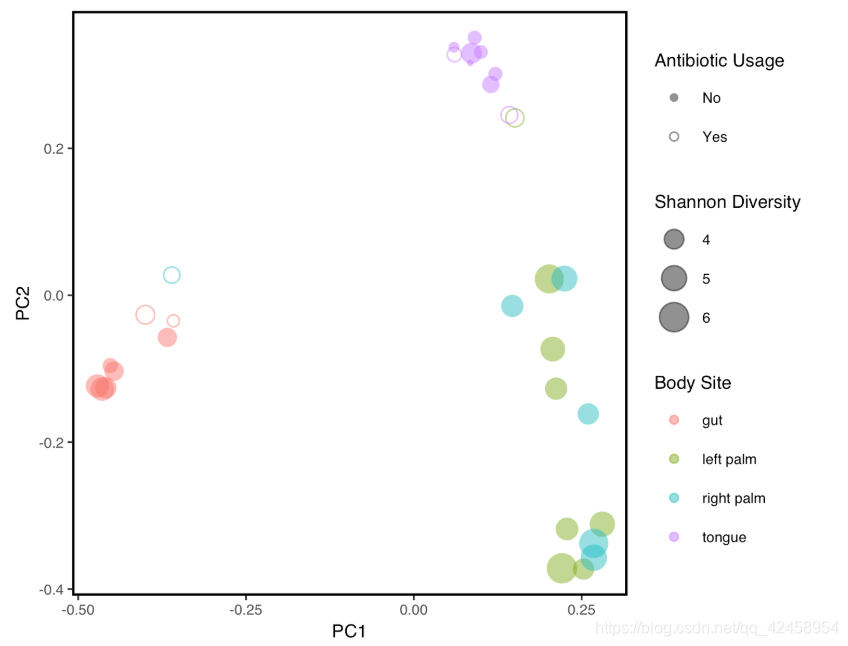
Heatmap
library(tidyverse)
library(qiime2R)
metadata<-read_q2metadata("sample-metadata.tsv")
SVs<-read_qza("table.qza")$data
taxonomy<-read_qza("taxonomy.qza")$data %>% parse_taxonomy()
taxasums<-summarize_taxa(SVs, taxonomy)$Genus
taxa_heatmap(taxasums, metadata, "body-site")
ggsave("heatmap.pdf", height=4, width=8, device="pdf") # save a PDF 4 inches by 8 inches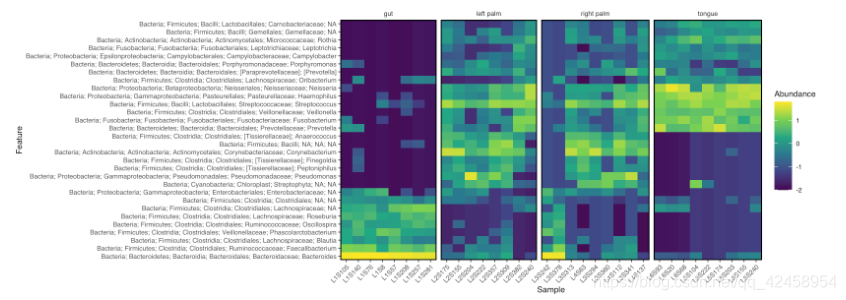
物种分类柱状图的绘制
library(tidyverse)
library(qiime2R)
metadata<-read_q2metadata("sample-metadata.tsv")
SVs<-read_qza("table.qza")$data
taxonomy<-read_qza("taxonomy.qza")$data %>% parse_taxonomy()
taxasums<-summarize_taxa(SVs, taxonomy)$Genus
taxa_barplot(taxasums, metadata, "body-site")
ggsave("barplot.pdf", height=4, width=8, device="pdf") # save a PDF 4 inches by 8 inches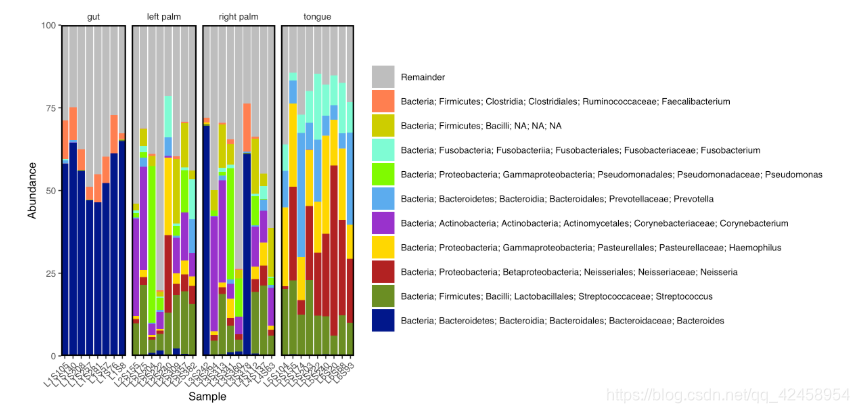
差异性丰度计算
library(tidyverse)
library(qiime2R)
library(ggrepel) # for offset labels
library(ggtree) # for visualizing phylogenetic trees
library(ape) # for manipulating phylogenetic trees
metadata<-read_q2metadata("sample-metadata.tsv")
SVs<-read_qza("table.qza")$data
results<-read_qza("differentials.qza")$data
taxonomy<-read_qza("taxonomy.qza")$data
tree<-read_qza("rooted-tree.qza")$data火山图
results %>%
left_join(taxonomy) %>%
mutate(Significant=if_else(we.eBH<0.1,TRUE, FALSE)) %>%
mutate(Taxon=as.character(Taxon)) %>%
mutate(TaxonToPrint=if_else(we.eBH<0.1, Taxon, "")) %>% #only provide a label to signifcant results
ggplot(aes(x=diff.btw, y=-log10(we.ep), color=Significant, label=TaxonToPrint)) +
geom_text_repel(size=1, nudge_y=0.05) +
geom_point(alpha=0.6, shape=16) +
theme_q2r() +
xlab("log2(fold change)") +
ylab("-log10(P-value)") +
theme(legend.position="none") +
scale_color_manual(values=c("black","red"))
ggsave("volcano.pdf", height=3, width=3, device="pdf") 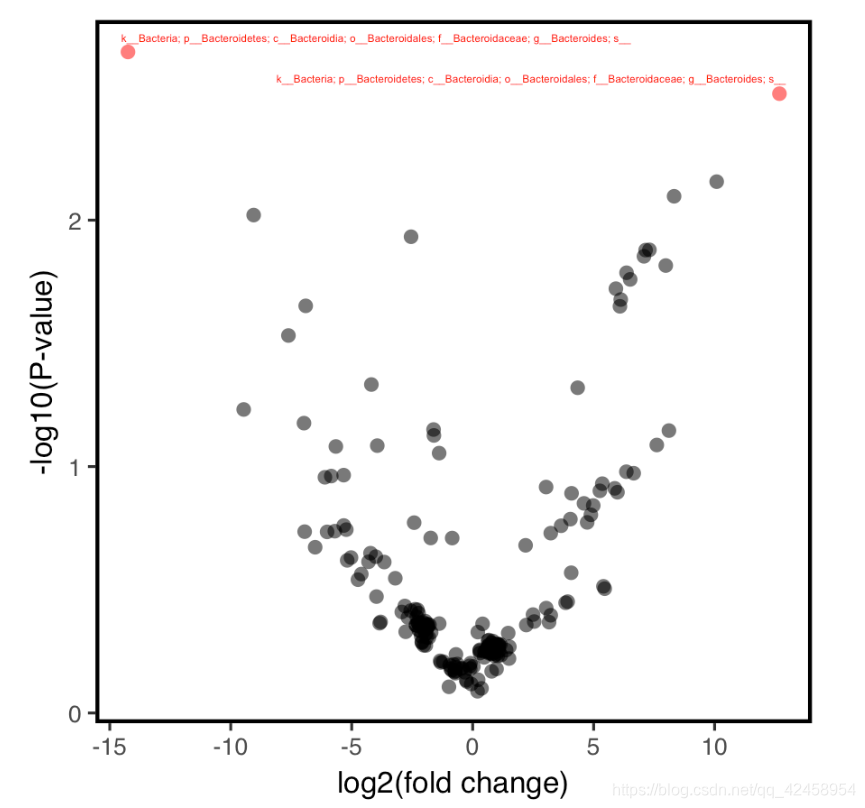
单个feature丰度图
clr<-apply(log2(SVs+0.5), 2, function(x) x-mean(x))
clr %>%
as.data.frame() %>%
rownames_to_column("Feature.ID") %>%
gather(-Feature.ID, key=SampleID, value=CLR) %>%
filter(Feature.ID=="4b5eeb300368260019c1fbc7a3c718fc") %>%
left_join(metadata) %>%
filter(`body-site`=="gut") %>%
ggplot(aes(x=subject, y=CLR, fill=subject)) +
stat_summary(geom="bar", color="black") +
geom_jitter(width=0.2, height=0, shape=21) +
theme_q2r() +
theme(legend.position="none")
ggsave("aldexbar.pdf", height=2, width=1.5, device="pdf") 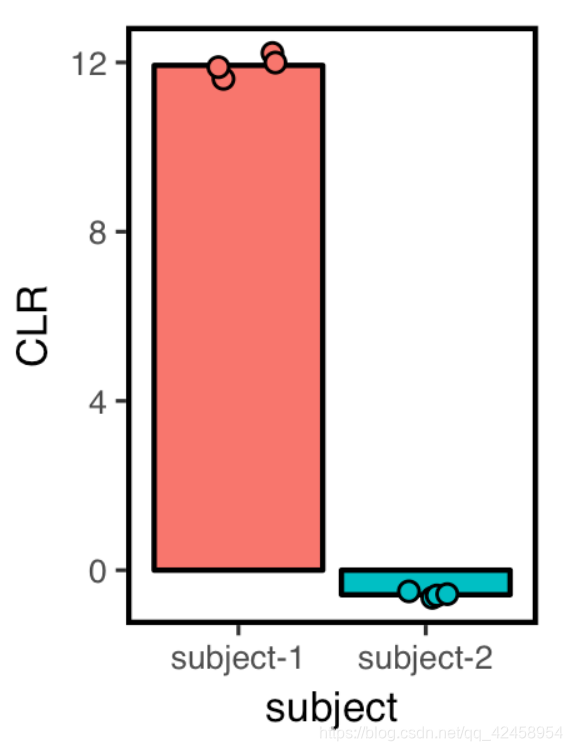
进化树绘制
results<-results %>% mutate(Significant=if_else(we.eBH<0.1,"*", ""))
tree<-drop.tip(tree, tree$tip.label[!tree$tip.label %in% results$Feature.ID]) # remove all the features from the tree we do not have data for
ggtree(tree, layout="circular") %<+% results +
geom_tippoint(aes(fill=diff.btw), shape=21, color="grey50") +
geom_tiplab2(aes(label=Significant), size=10) +
scale_fill_gradient2(low="darkblue",high="darkred", midpoint = 0, mid="white", name="log2(fold-change") +
theme(legend.position="right")
ggsave("tree.pdf", height=10, width=10, device="pdf", useDingbats=F) 


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









