






一、任务介绍
最近在做一个基因补全(impution)的任务,使用的是千人基因组计划(1KGP_30x_GRCh38)的数据,这是目前为止最为精确的人类基因测序的基因序列数据。我所做的任务需要对基因突变位点进行过滤:保留SNP类型,除去SV类型。
二、过滤工具
GATK是一个十分方便的工具,可以直接安装到conda环境中,并且根据需求对vcf文件进行处理。关于我自己的过滤任务,我所使用到的指令是SelectVariant中的option,指令内容大致为:
gatk SelectVariants -V input.vcf --select-type-to-include SNP -O output.vcf
指令内容简单易懂,我就不具体解释了
三、报错内容以及解决方式
第一次尝试过滤vcf文件的时候,我使用的是1KGP_phase3中22号染色体的数据(vcf格式),第一次是可以正常筛选的。
在我使用1KGP_30x_GRCh38做相同操作的时候出现了以下报错: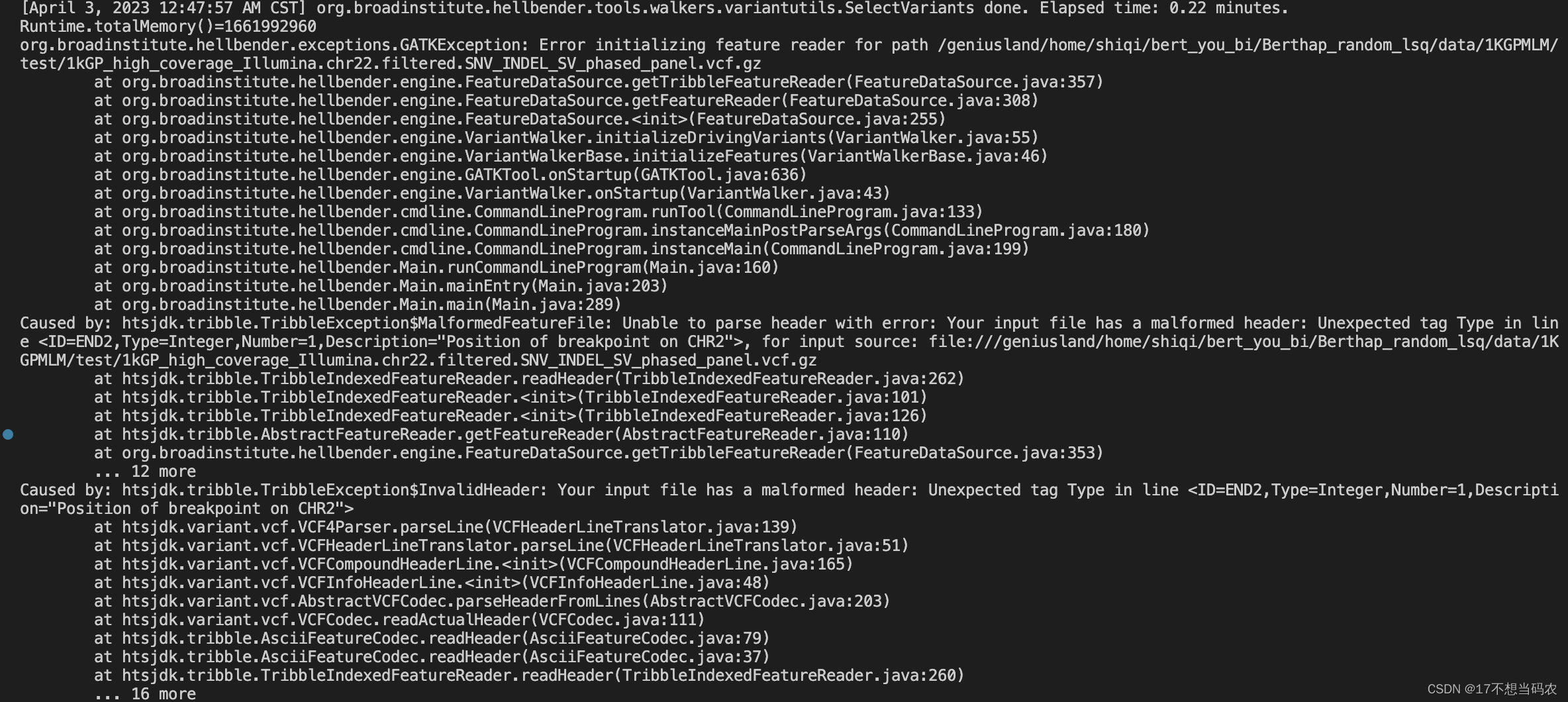
其中报错的关键信息是:Unable to parse header with error: Your input file has a malformed header: Unexpected tag Type in line <ID=END2,Type=Integer,Number=1,Description=“Position of breakpoint on CHR2”>
大概意思就是,gatk在处理vcf文件的时候在读取头信息的时候出现了预期之外的一些标签属性。我尝试换数据、更换指令,等等都没有改善该情况。然后我发现了一篇文章和我一模一样的问题(附上链接:https://github.com/samtools/hts-specs/issues/642)
文章大概内容的意思就是,头信息中有多个tag,例如这条信息里面就是ID、Type、Number等等,他们的排列顺序是有一定规则的。
文章中有人给出了回答:
##INFO=`<ID=ID,Number=number,Type=type,Description="description",Source="description",Version="128">`
和我所处理的vcf文件中tag的顺序是有出入的(Number和Type的顺序是反了),所以我尝试更换一下vcf文件中tag的顺序通过以下指令:
sed -i ‘s/ID=END2,Type=\([^,]*\),Number=\([^,]*\)/ID=END2,Number=\2,Type=\1/g' inputfile.vcf
在调换过顺序后重新运行了一下筛选指令,发现可以运行了!
简单吐槽一嘴:下载的是官方给的数据,这里面也能有错误也是绝了…















 2万+
2万+











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









