






参考文献:A tutorial on conducting genome-wide association studies: Quality control and statistical analysis
(45条消息) 全基因组关联分析学习资料(GWAS tutorial)20210313更新版_gwas summary下载_橙子牛奶糖的博客-CSDN博客
 https://github.com/MareesAT/GWA_tutorial
https://github.com/MareesAT/GWA_tutorial一. 前言
1. GWAS的目的:在全基因组范围找到与特定表型相关的遗传变异(SNP)
(计算SNP等位基因频率与表型的线性关联)
2.数据处理
(1)预处理:基因型-表型-插补
(2)进阶分析:
群体分层,population stratification
不同队列的重复验证,replication
LocusZoom---regional association plots
相似条件分析-approximate conditional analysis(去除lead SNP后在跑一次GWAS关联分析,找到更多有强关联的信号)
LD score regression-表型间的遗传相关性。(由基因型产生的相关性)
基因富集分析(与leadSNP相关基因 + 与leadSNP的LD r2>0.8 SNP的基因)
层次聚类分析(表型相关的lead SNPs与其它表型的相关性)
蛋白-蛋白互作网络(哪些蛋白共同调控了表型)
point of contact analysis(哪些位点导致表型间有相关性)
二. 质控
1. linux + plink2
+ 表型.txt,协变量.txt--------plink------> 关联分析
2. 质控内容:
(1)个体和SNPs缺失(0.2-0.02);--geno --mind
(2)性别统计错误(X纯度>0.8,<0.2);-check-sex
个体性别记录与真实性别的差异----根据X染色体杂合率/纯合率检验;男性的X染色体纯合度>0.8,女性<0.2
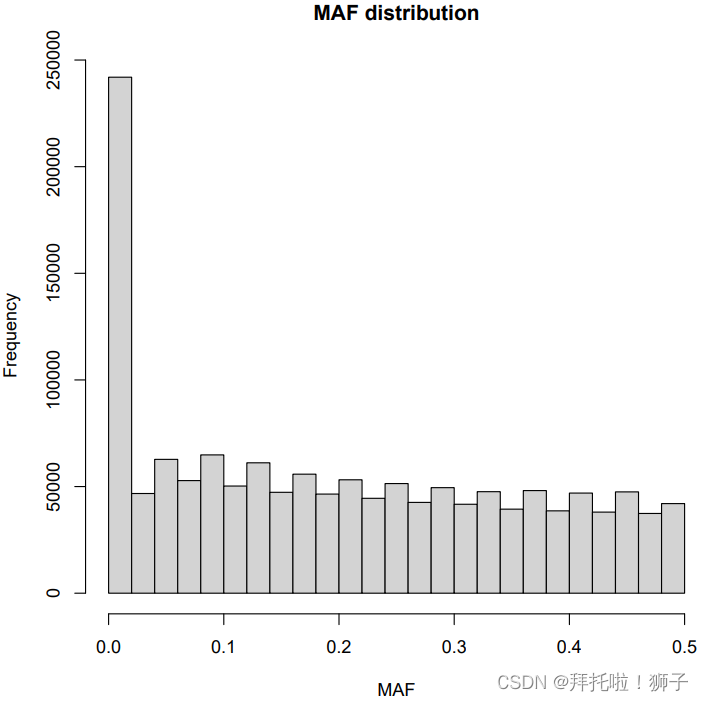
(3)MAF(0.01-0.05);--maf
低MAF的SNP很难检测出与表型的关联--除非是超大样本
(4)哈代-温伯格平衡检验(1e-10,1e-6);
如果HW不平衡,可能是由于群体分层,基因分型错误等。
(5)个体杂合度(u+-3SD);
过高或过低---样本污染,近亲繁殖
(6)相关性(亲缘关系pi-hat: 0.2);--genome --min
计算所有样本对的IBD(identity of descent)
(7)种群分层;--genome --cluster --mds-plot k
可以用PCA结果作为协变量来控制
conda activate r4.2
conda install plink
##提供的数据信息:表型(结局)为二值变量,种族同质(北欧和西欧)
plink --bfile HapMap_3_r3_1 --missing
##文本文件用--file,二进制文件用--bfile(1)个体和SNP缺失(先过滤SNP,再过滤个体)
# Delete SNPs with missingness >0.2.
plink --bfile HapMap_3_r3_1 --geno 0.2 --make-bed --out HapMap_3_r3_2
# Delete individuals with missingness >0.2.
plink --bfile HapMap_3_r3_2 --mind 0.2 --make-bed --out HapMap_3_r3_3
# Delete SNPs with missingness >0.02.
plink --bfile HapMap_3_r3_3 --geno 0.02 --make-bed --out HapMap_3_r3_4
# Delete individuals with missingness >0.02.
plink --bfile HapMap_3_r3_4 --mind 0.02 --make-bed --out HapMap_3_r3_5
##如果--geno 和 --mind放在同一行,
##表示去除原始样本中高缺失率的SNPs,以及去除原始样本中高缺失率的个体。
##但分开进行的意思是:在去除SNPs的数据基础上,再去除个体。直接使用较低的阈值(如0.02)进行处理,相比先使用较高的阈值(0.2)再用较低的阈值(0.02)更为严格。将剔除更多缺失的SNP和个体,导致数据集变小,样本和位点数量减少,影响后续的分析结果和解释。
wc -l HapMap_3_r3_1.bim
#1457897 HapMap_3_r3_1.bim
wc -l HapMap_3_r3_5.bim
#1430443 HapMap_3_r3_5.bim
wc -l HapMap_3_r3_1.fam
#165 HapMap_3_r3_1.fam
wc -l HapMap_3_r3_5.fam
#165 HapMap_3_r3_5.fam
(2)Check for sex discrepancy;
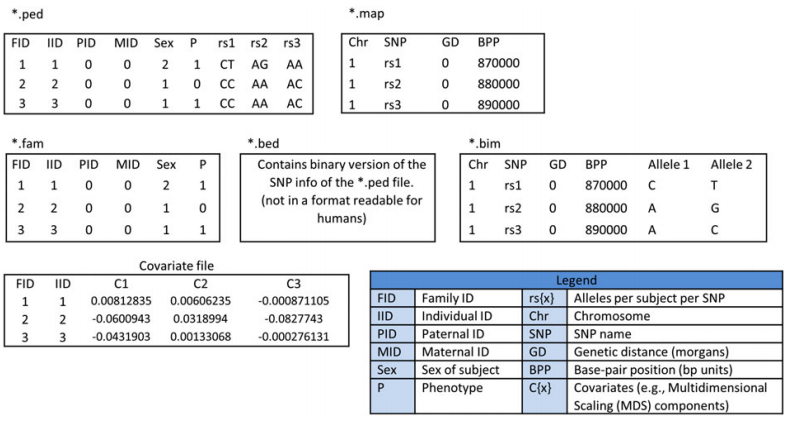
F:X染色体杂合率/纯合率,男性接近1,女性接近0.--check-sex [女性阈值] [男性阈值] 默认的参数为 0.2 0.8
plink --bfile HapMap_3_r3_5 --check-sex gwas/1_QC_GWAS/plink.sexcheck;

第3列为PED文件中的性别,第4列为通过X染色体估计的性别,第五列表示是否一致,不一致时会表示为PROBLEM,第六列是估计的F系数。
grep "PROBLEM" plink.sexcheck| awk '{print$1,$2}'> sex_discrepancy.txt
plink --bfile HapMap_3_r3_5 --remove sex_discrepancy.txt --make-bed --out HapMap_3_r3_6
#删除该个体
#plink --bfile HapMap_3_r3_5 --impute-sex --make-bed --out HapMap_3_r3_6
#基于基因型信息的性别进行插补(3)MAF
#先去除了chr1-22之外的染色体SNPs
awk '{ if ($1 >= 1 && $1 <= 22) print $2 }' HapMap_3_r3_6.bim > snp_1_22.txt
plink --bfile HapMap_3_r3_6 --extract snp_1_22.txt --make-bed --out HapMap_3_r3_7
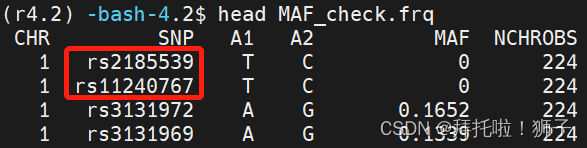
plink --bfile HapMap_3_r3_7 --freq --out MAF_check
plink --bfile HapMap_3_r3_7 --maf 0.05 --make-bed --out HapMap_3_r3_8
(4)HWE
对每个SNP进行独立的检验,并计算其p值。如果在一个群体中,某个SNP的基因型频率与哈代-温伯格平衡的预期频率有显著差异,就可能表明该SNP存在非随机的基因型分布,可能与选择、漂移或其他进化过程有关。
plink --bfile HapMap_3_r3_8 --hwe 1e-6 --make-bed --out HapMap_hwe_filter_step1
#--hwe默认只去除control(参考SNP)中<1e-6的
plink --bfile HapMap_hwe_filter_step1 --hwe 1e-10 --hwe-all --make-bed --out HapMap_3_r3_9
#然后再用1e-10的阈值将case中极度偏离的SNP去除。  没有偏离HWE的SNPs
没有偏离HWE的SNPs
(5)去除杂合率偏离其平均值大于3sd的个体
杂合性检查是在一组不高度相关的snp上进行的。因此,需要先排除LD-SNP
plink --bfile HapMap_3_r3_9 --exclude inversion.txt --range --indep-pairwise 50 5 0.2 --out indepSNP
#window size=50,shift step=5个SNPs,该SNP对其他SNPs回归的多重相关系数=0.2;
plink --bfile HapMap_3_r3_9 --extract indepSNP.prune.in --het --out R_check
#104144 variants and 164 people pass filters and QC.--het 可以通过距估计来估计F系数:定义为
(观测纯合基因型个数O - 预期纯合基因型个数E)/(总基因型数N - 预期纯合基因型个数E)
1_QC_GWAS/R_check.het 
(我也不知道为什么不直接用F过滤?????算它的意义何在????)
het <- read.table("R_check.het", head=TRUE)
het$HET_RATE = (het$"N.NM." - het$"O.HOM.")/het$"N.NM." ##即观测杂合/总基因型数
het_fail = subset(het, (het$HET_RATE < mean(het$HET_RATE)-3*sd(het$HET_RATE)) | (het$HET_RATE > mean(het$HET_RATE)+3*sd(het$HET_RATE)));
het_fail$HET_DST = (het_fail$HET_RATE-mean(het$HET_RATE))/sd(het$HET_RATE);
#z-score归一化
write.table(het_fail, "fail-het-qc.txt", row.names=FALSE) 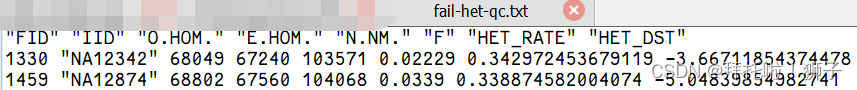
sed 's/"// g' fail-het-qc.txt | awk '{print$1, $2}'> het_fail_ind.txt
#去除",只提取第1,2列 个体信息![]() --即需要去除的杂合率极端的个体。
--即需要去除的杂合率极端的个体。
plink --bfile HapMap_3_r3_9 --remove het_fail_ind.txt --make-bed --out HapMap_3_r3_10
#去除后剩余162个有表型的个体(6)亲缘关系
PLINK中使用 PI_HAT 值(--genome)来推定IBD(identity by decent)的值。该方法基于HMM,通过矩估计(method-of-moments)来计算 IBD=1, 2或0 的概率。
PI_HAT:为IBD比例 , 即 P(IBD=2) + 0.5*P(IBD=1),PI_HAT的值与对应关系如下所示:
- PI_HAT=0 无亲缘关系
- PI_HAT=0.25 表兄弟
- PI_HAT=0.5 亲子或兄弟姐妹
- PI_HAT=1 本人或同卵双胞胎
plink --bfile HapMap_3_r3_10 --filter-founders --make-bed --out HapMap_3_r3_11
## founders (individuals without parents in the dataset).
##剩下110个有表型的个体
plink --bfile HapMap_3_r3_11 --extract indepSNP.prune.in --genome --min 0.2 --out pihat_min0.2_in_founders

看哪个个体缺失的数据多,就去掉它
plink --bfile HapMap_3_r3_11 --missing
#plink.imissMISS_PHENO:是否表型缺失;N_MISS:缺失的SNP数目;N_GENO:总SNP数;F_MISS:缺失率.(注意是找对应的IID,而不是FID)
13291 NA07045 N 2552 1073226 0.002378
1454 NA12813 N 1947 1073226 0.001814
In our dataset the individual 13291 NA07045 had the lower call rate.
vi 0.2_low_call_rate_pihat.txt
i
13291 NA07045
# Press esc on keyboard!
:x## 删除该pi_hat>0.2即有亲缘关系个体中,F_MISS即SNPs缺失率更高的个体
plink --bfile HapMap_3_r3_11 --remove 0.2_low_call_rate_pihat.txt --make-bed --out HapMap_3_r3_12next step using:
# - The bfile HapMap_3_r3_12
# - indepSNP.prune.in
三. 种群分层控制-----PCA 或 MDS
----plink 和 EIGENSTRAT
1. PCA
不同群体SNP频率不同,关联假阳性,对群体做PCA分析,取PCA结果eigenvec($loadings)作为协变量加入关联分析中。
为什么用$loadings(特征向量),而不是$scores(变量投影在该PC的位置)??
loadings值反映了原始变量(遗传变量SNP)在主成分上的权重或相关性。较大的loadings值表示对应的遗传变异在主成分中具有更高的权重和重要性。这意味着这些遗传变异在样本之间的差异方面起着更大的作用,可以用来划分群体或亚群体。识别在主成分中对样本之间遗传差异贡献最大的遗传变异。这有助于确定主成分所代表的遗传结构,从而在群体分层中控制。
scores相对位置更多地关注变量在主成分方向上的表现或位置,而不是对群体结构的贡献。
计算膨胀系数(Calculating Genomic Inflation Factor):基因组膨胀因子λ定义为得到的卡方检验统计量的中值除以卡方分布的预期中值。预期的P值膨胀系数为1,当实际膨胀系数越偏离1,说明存在群体分层的现象越严重,需重新矫正群体分层。
lambda = median(z^2, na.rm = TRUE)/qchisq(0.5, 1)当群体出现分层时,常规手段就是将分层的群体独立分析,最后再做meta分析。
2. plink中的方法:多维缩放(multimensional scaling,MDS)
默认使用一对个体间的共享的等位基因占全基因组的比例,即基因型的相似性(genomic relationship)来计算样本之间的相似性矩阵,然后进行特征值分解,将选择的主成分与原始数据的相似性矩阵进行矩阵相乘,得到样本在低维空间中的坐标。
可以绘制被调查样本的MDS和已知种族结构的群体(HapMap/1KG数据),来确定其样本的种族信息,以及样本生成的成分分数偏离样本目标群体的个体。
*******与PCA的区别:
输入数据类型(协方差矩阵 vs. 相似性/距离矩阵)和降维目标(最大方差解释 vs. 保持相对距离关系)
PCA旨在找到数据的主要变化方向,以最大程度地解释数据的方差。经典MDS旨在保持样本之间的相对距离关系,并将样本映射到低维空间中。
接下来会用1000 Genomes Project来检查HapMap_3_r3_12的群体分层情况。non-European ethnic background 的个体会被移除。以及生成一个协变量文件,它有助于调整欧洲受试者中剩余的人口分层。
cd ../2_Population_stratification
cp ../1_QC_GWAS/HapMap_3_r3_12.* ./
cp ../1_QC_GWAS/indepSNP.prune.in ./##Download 1000 Genomes data ##
#contains genetic data of 629 individuals from different ethnic backgrounds.
nohup wget ftp://ftp-trace.ncbi.nih.gov/1000genomes/ftp/release/20100804/ALL.2of4intersection.20100804.genotypes.vcf.gz &
plink --vcf ALL.2of4intersection.20100804.genotypes.vcf.gz --make-bed --out ALL.2of4intersection.20100804.genotypes
##文件中缺乏rs-identifiers,它是用.表示的
plink --bfile ALL.2of4intersection.20100804.genotypes --set-missing-var-ids @:#[b37]\$1,\$2 --make-bed --out ALL.2of4intersection.20100804.genotypes_no_missing_IDs
##设置缺失的rs标识符,@和#占位符,[b37]表示使用b37(GRCh37)基因组版本的参考坐标,$1和$2表示将原始的第一列(chr)和第二列(pos)作为新的标识符。
#\$1 和 \$2 的目的是将 $1 和 $2 视为纯文本而不是变量,并将其传递给 PLINK 命令的相应参数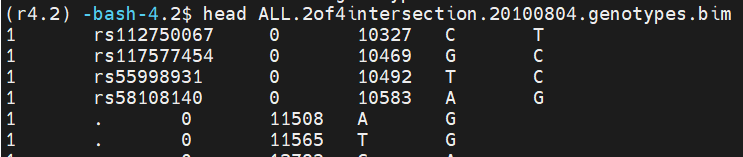
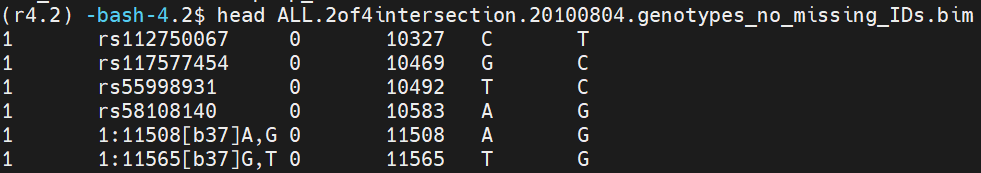
### 对1kG数据进行相同的质控
plink --bfile ALL.2of4intersection.20100804.genotypes_no_missing_IDs --geno 0.2 --allow-no-sex --make-bed --out 1kG_MDS
plink --bfile 1kG_MDS --mind 0.2 --allow-no-sex --make-bed --out 1kG_MDS2
plink --bfile 1kG_MDS2 --geno 0.02 --allow-no-sex --make-bed --out 1kG_MDS3
plink --bfile 1kG_MDS3 --mind 0.02 --allow-no-sex --make-bed --out 1kG_MDS4
plink --bfile 1kG_MDS4 --maf 0.05 --allow-no-sex --make-bed --out 1kG_MDS5
# 从1kG中提取出HapMap的SNP
awk '{print$2}' HapMap_3_r3_12.bim > HapMap_SNPs.txt
plink --bfile 1kG_MDS5 --extract HapMap_SNPs.txt --make-bed --out 1kG_MDS6
#1000993 1kG_MDS6.bim
#从HapMap中提取出1kG的SNP
awk '{print$2}' 1kG_MDS6.bim > 1kG_MDS6_SNPs.txt
plink --bfile HapMap_3_r3_12 --extract 1kG_MDS6_SNPs.txt --recode --make-bed --out HapMap_MDS
#1000993 HapMap_MDS.bim
#--recode 将输出文件二进制bed以重新编码的格式生成PED和MAP文件
#awk 'FNR==NR {a[$2]; next} $1 in a' 1kG_MDS5.bim HapMap_SNPs.txt|wc -l
#1000993 只是两文件中rs相同的SNPs
#FNR==NR 的判断条件表示当前处理的是第一个文件,而 FNR!=NR 表示当前处理的是非第一个文件
#{a[$2]; next}表示将文件1kG_MDS5.bim中的第二列内容存储到数组a中,并继续处理下一行。
#$2 in a 表示判断文件HapMap_SNPs.txt中的第二列内容是否存在于数组a中,如果存在,则打印该行。
awk '{print$2,$4}' HapMap_MDS.map > buildhapmap.txt # rs + pos
plink --bfile 1kG_MDS6 --update-map buildhapmap.txt --make-bed --out 1kG_MDS7
将输入数据集 1kG_MDS6 中的位点信息根据 buildhapmap.txt 中提供的新信息进行更新 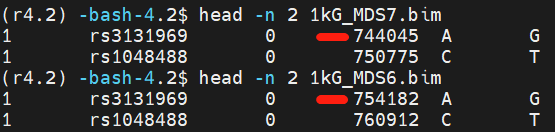

按照HapMap位点信息更新完的rs3131969(1kG_MDS7.bim)的position,反而和NCBI中rs3131969的位置不匹配,hhhh
#### 对1kG数据和HapMap数据进行合并
先要确定两数据中的SNP信息是否完全一致,先定义参考基因型,SNP不一致可能是由于strand的问题(链性问题通常指的是基因型数据和参考基因组之间链性【正负链】的对应关系不一致)(即两文件参考基因型一致,位置一致的SNP,基因型不一致$6,可能分别来源于正、负链),将这部分进行flip翻转;然后其它原因导致不同的,进行删除。
awk '{print$2,$5}' 1kG_MDS7.bim > 1kg_ref-list.txt
plink --bfile HapMap_MDS --reference-allele 1kg_ref-list.txt --make-bed --out HapMap-adj
#same reference genome for all SNPs.
awk '{print$2,$5,$6}' 1kG_MDS7.bim > 1kGMDS7_tmp
awk '{print$2,$5,$6}' HapMap-adj.bim > HapMap-adj_tmp
sort 1kGMDS7_tmp HapMap-adj_tmp |uniq -u > all_differences.txt
#排序合并,将只出现在一个文件中的输出
# 1624
awk '{print$1}' all_differences.txt | sort -u > flip_list.txt
# 812 SNPs 去除了SNPs的rs号相同的
plink --bfile HapMap-adj --flip flip_list.txt --reference-allele 1kg_ref-list.txt --make-bed --out corrected_hapmap
awk '{print$2,$5,$6}' corrected_hapmap.bim > corrected_hapmap_tmp
sort 1kGMDS7_tmp corrected_hapmap_tmp |uniq -u > uncorresponding_SNPs.txt
# 84,进行链翻转flip后,还有84个SNPs不同,直接删去
awk '{print$1}' uncorresponding_SNPs.txt | sort -u > SNPs_for_exlusion.txt
# 42
plink --bfile corrected_hapmap --exclude SNPs_for_exlusion.txt --make-bed --out HapMap_MDS2
plink --bfile 1kG_MDS7 --exclude SNPs_for_exlusion.txt --make-bed --out 1kG_MDS8
##合并
plink --bfile HapMap_MDS2 --bmerge 1kG_MDS8.bed 1kG_MDS8.bim 1kG_MDS8.fam --allow-no-sex --make-bed --out MDS_merge2
plink --bfile MDS_merge2 --extract indepSNP.prune.in --genome --out MDS_merge2
plink --bfile MDS_merge2 --read-genome MDS_merge2.genome --cluster --mds-plot 10 --out MDS_merge2
#计算每对个体之间的共享等位基因数量,并根据其比例估计亲缘关系。结果以两个个体之间的PI_HAT值表示[0,2],0无亲缘关系
##


#下载1kG的种群信息
wget ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/release/20100804/20100804.ALL.panel
awk '{print$1,$1,$2}' 20100804.ALL.panel > race_1kG.txt
sed 's/JPT/ASN/g' race_1kG.txt>race_1kG2.txt
sed 's/ASW/AFR/g' race_1kG2.txt>race_1kG3.txt
sed 's/CEU/EUR/g' race_1kG3.txt>race_1kG4.txt
sed 's/CHB/ASN/g' race_1kG4.txt>race_1kG5.txt
sed 's/CHD/ASN/g' race_1kG5.txt>race_1kG6.txt
sed 's/YRI/AFR/g' race_1kG6.txt>race_1kG7.txt
sed 's/LWK/AFR/g' race_1kG7.txt>race_1kG8.txt
sed 's/TSI/EUR/g' race_1kG8.txt>race_1kG9.txt
sed 's/MXL/AMR/g' race_1kG9.txt>race_1kG10.txt
sed 's/GBR/EUR/g' race_1kG10.txt>race_1kG11.txt
sed 's/FIN/EUR/g' race_1kG11.txt>race_1kG12.txt
sed 's/CHS/ASN/g' race_1kG12.txt>race_1kG13.txt
sed 's/PUR/AMR/g' race_1kG13.txt>race_1kG14.txt
awk '{print$1,$2,"OWN"}' HapMap_MDS.fam>racefile_own.txt
cat race_1kG14.txt racefile_own.txt | sed -e '1i\FID IID race' > racefile.txt
#i即在第一行前添加,d是删除

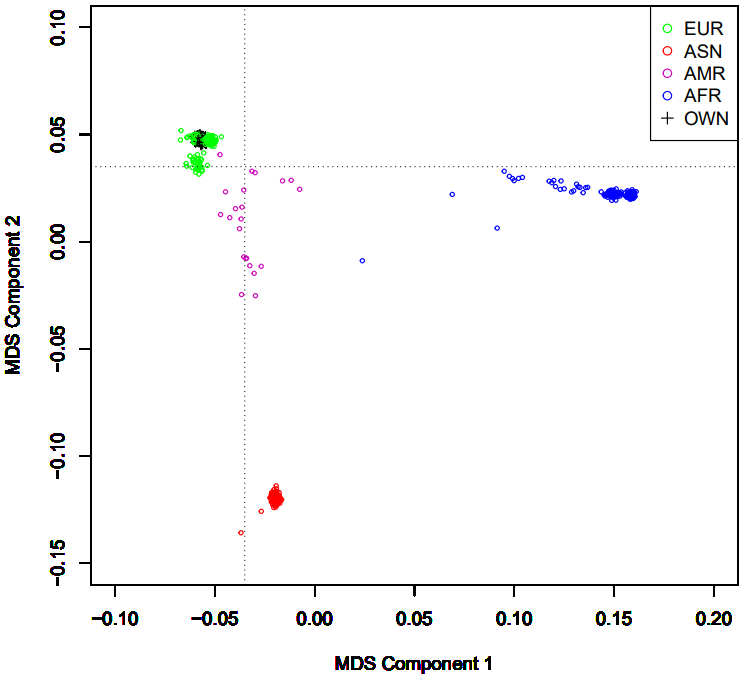 数据嵌入在1kG的EUR中无离群个体
数据嵌入在1kG的EUR中无离群个体
##当有离群点时 ## Exclude ethnic outliers
为了排除异常值,需要围绕感兴趣的race_group设置阈值(而非一个fixed阈值)
#为EUR设置阈值 $4即C1,$5即C2,也就是MDS图的横纵坐标。$1,$2即FID和IID
awk '{ if ($4 <-0.04 && $5 >0.03) print $1,$2 }' MDS_merge2.mds > EUR_MDS_merge2
plink --bfile HapMap_3_r3_12 --keep EUR_MDS_merge2 --make-bed --out HapMap_3_r3_13
# outliers individuals would have been removed
## Create covariates based on MDS
The values of the 10 MDS dimensions are subsequently used as covariates in the association analysis。C1-C10会作为后续关联分析的协方差,进行控制。
plink --bfile HapMap_3_r3_13 --extract indepSNP.prune.in --genome --out HapMap_3_r3_13
plink --bfile HapMap_3_r3_13 --read-genome HapMap_3_r3_13.genome --cluster --mds-plot 10 --out HapMap_3_r3_13_mds
##去除完outliers个体之后,重新计算了个体间的亲缘系数和MSD
awk '{print$1, $2, $4, $5, $6, $7, $8, $9, $10, $11, $12, $13}' HapMap_3_r3_13_mds.mds > covar_mds.txt
#adjust for remaining population stratification得到文件:- HapMap_3_r3_13 ,- covar_mds.txt
四. 关联分析(二元性状 & 数量性状)
1. 二元结局
在PLINK中,SNP与二元结果之间的关联(1代表未受影响,2代表受影响;0和-9代表缺失)
PLINK中--assoc选项执行的X2关联测试是不包含协变量的。使用--logistic选项将执行包含协变量的逻辑回归分析。
2. 数量性状结局
--assoc选项将通过执行常用的Student's t检验的渐进版本来比较两个均值。这个选项不允许使用协变量。P--linear选项以每个单独的SNP作为预测变量执行线性回归分析。--linear选项允许使用协变量。
3. 多重假设检验矫正
Bonferroni校正、Benjamini-Hochberg错误发现率(FDR)、置换(permutation)
# assoc
# plink --bfile HapMap_3_r3_13 --assoc --out assoc_results #不能校正协方差
# logistic
plink --bfile HapMap_3_r3_13 --covar covar_mds.txt --logistic --hide-covar --out logistic_results
awk '!/'NA'/' logistic_results.assoc.logistic > logistic_results.assoc_2.logistic
#移除输出中的na值
#如果是数量性状,--logistic可以替换成 --linear
#多重假设检验校正
plink --bfile HapMap_3_r3_13 -assoc --adjust --out adjusted_assoc_results
#结果会有Bonferroni corrected p-value, FDR and others
##permutation
plink --bfile HapMap_subset_for_perm --assoc --mperm 1000000 --out subset_1M_perm_result
sort -gk 4 subset_1M_perm_result.assoc.mperm > sorted_subset.txt #按p值排序
##QQ-plot和manhattan
qq(results$P, main = "Q-Q plot of GWAS p-values : log")
manhattan(results,chr="CHR",bp="BP",p="P",snp="SNP", main = "Manhattan plot: logistic")
五. PRS 多基因风险评分
很多对表型有贡献的位点达不到5e-8会被过滤,单个位点对表型的解释度很低。
PRS就是综合了更多对表型有贡献的位点,算出来的效应值(风险得分)。
将个体的多个SNP的影响大小组合成一个可以用来预测疾病风险的汇总评分(个体水平)
(根据一个人携带的风险变异的数量来计算,根据个体携带的风险等位基因数量乘以性状特异性权重,即SNP效应大小【GWAS关联分析结果】加权,然后对所有风险基因求和,来计算每个人的 PRS。)
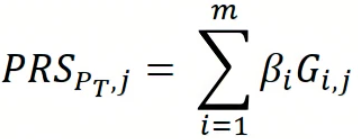
PT表示选择的p值的阈值;
i表示符合该阈值下的SNP,i = 1, 2, ..., m;
βi表示SNP的效应值,线性表型,该值为β;二元表型,该值为OR;
Gi,j 表示SNP的基因型,分别用{0,1,2}表示;

一旦计算了个体关于某个性状表型的PRS,就可以在回归分析中,使用这些分数来预测预期的表型。预测精度可以用R2度量。即PRS可以解释表型差异的比例。
R = cov(X, Y) / (σX * σY)
(由于X,Y相关性导致XY方差 的比例)
R通常指pearson相关系数,用于衡量因变量与自变量之间的线性相关程度。它的取值范围在-1到1之间,表示相关性的方向和强度。
R²是对相关系数R的平方,表示自变量对因变量变异性的解释程度(因变量的变异中可以被自变量解释的比例)。它的取值范围在0到1之间,R²的值越接近1,表示自变量能够解释更大比例的因变量变异,拟合效果越好;R²只能度量线性关系的解释程度,对于非线性关系可能不准确。
PRS的预测准确性主要取决于分析性状的(共)遗传力、SNP的数量,发现样本的大小。目标样本的大小只影响R2的可靠性,通常几千名受试者足以达到显著的 R 2。
wget https://github.com/choishingwan/PRSice/releases/download/2.1.11/PRSice_linux.zip
unzip PRSice_linux.zip
##用zip中给的示例文件来练习
## 二元
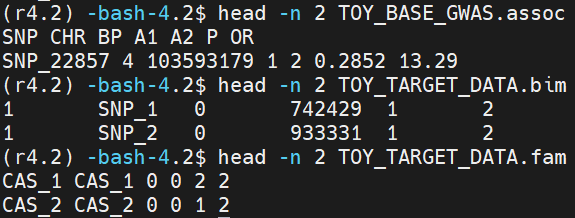
Rscript PRSice.R --dir . --prsice ./PRSice_linux --base TOY_BASE_GWAS.assoc
--target TOY_TARGET_DATA --thread 1 --stat OR --binary-target T
# base:summary statistics from the base sample发现样本,最少要包括effect size(或OR)和p
# target:plink基因型数据的前缀
# PRSice默认先clump最相关的SNPs(最低p值),忽略其他SNPs
## 数量性状
Rscript PRSice.R --dir . --prsice ./PRSice_linux --base TOY_BASE_GWAS.assoc
--target TOY_TARGET_DATA --thread 1 --stat BETA --beta --binary-target F
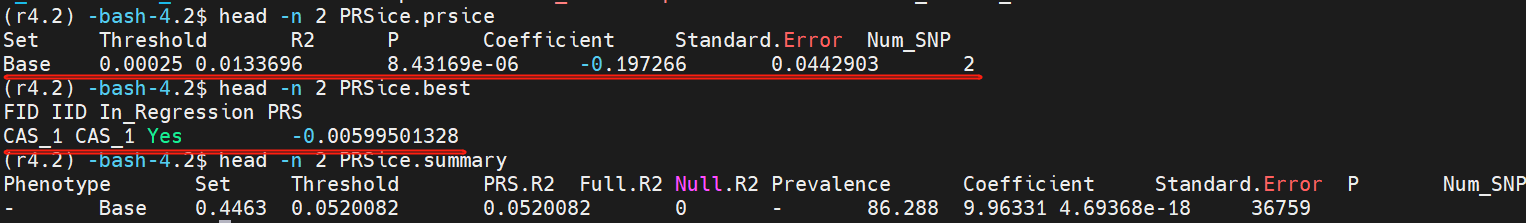
* PRSice.prsice文件包含:在给定不同阈值的P值以后,符合要求的SNP数量(Num_SNP),SNP的解释度(R2),回归系数。
* PRSice.best 计算的是每个个体最优的PRS值.
* PRSice.summary 即该表型下最优的模型。包含P的阈值,PRS的解释方差,所有变量的解释方差,协变量的解释方差,回归系数,P值,该阈值下的SNP数量。
R^2 = 1 - (SSR / SST)
其中,SSR是残差平方和(Sum of Squares of Residuals),表示模型预测值与实际观测值之间的差异的平方和。SST是总平方和(Total Sum of Squares),表示观测值与观测值均值之间的差异的平方和。
线性回归模型中,系数(即回归系数或斜率)表示自变量对因变量的影响程度。每个自变量都有一个对应的系数,用于衡量该自变量单位变化对因变量的平均影响。R2(R-squared)则是用来评估整个回归模型的拟合程度,即模型解释的方差比例。它表示因变量的变异有多少能够被模型解释。















 358
358

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









