







 本文详细描述了一次使用Gromacs进行的分子动力学模拟,涉及原子平板、蛋白质链与水分子的结构搭建,通过PBC=xy的边界条件、离子平衡和NVT热力学平衡,最终实现电中性系统的构建与电导率计算。关键步骤包括结构生成、参数设置、能量最小化和温度控制等。
本文详细描述了一次使用Gromacs进行的分子动力学模拟,涉及原子平板、蛋白质链与水分子的结构搭建,通过PBC=xy的边界条件、离子平衡和NVT热力学平衡,最终实现电中性系统的构建与电导率计算。关键步骤包括结构生成、参数设置、能量最小化和温度控制等。
这是一次pbc=xy时成功nvt平衡的例子,用两个蛋白和一个水层进行模拟。
做了两个相对原子质量为0.01(为了满足gromacs的仿真条件),电荷为0的原子组成的原子平板,为了满足pbc = xy的条件
用packmol搭建结构
gromacs自带的插入分子功能gmx insert-molecules能实现的用packmol都能实现。
two_level.inp
tolerance 1.0
filetype pdb
output two_level.pdb
structure chain_A.pdb
number 1
inside box 0. 0. 0. 60. 60. 60.
constrain_rotation x 0. 20.
constrain_rotation y 0. 20.
end structure
structure CentredWaterInSheets.pdb
number 1
inside box 0. 0. 60. 60. 60. 120.
end structure
structure chain_B.pdb
number 1
inside box 0. 0. 120. 60. 60. 180.
constrain_rotation x 0. 20.
constrain_rotation y 0. 20.
end structure
生成gro后更改盒子大小
gmx editconf -f two_level.pdb -o two_level.gro
然后在gro文件中的最后一行中修改盒子尺寸为
6.00000 6.00000 18.00000
经过测试,更改每个蛋白盒子之间的距离不会造成系统的崩溃
搭建top
有关链的itp文件是用pdb2gmx功能做的,然后加入到top中,gro和top不匹配的问题通过更改最后的分子顺序解决。
#include "oplsaa.ff/forcefield.itp"
#include "metal.itp"
#include "mPlus.itp"
#include "mMinus.itp"
#include "two_test_Protein_chain_A.itp"
#include "two_test_Protein_chain_B.itp"
#include "oplsaa.ff/spc.itp"
#include "oplsaa.ff/ions.itp"
#ifdef POSRES_WATER
; Position restraint for each water oxygen
[ position_restraints ]
; i funct fcx fcy fcz
1 1 1000 1000 1000
#endif
[ system ]
; Name
Water In Sheets and protein
[ molecules ]
; Compound #mols
Protein_chain_A 1
MP 360
SOL 3947
NA 31
NA 76
CL 76
MM 360
Protein_chain_B 1
[ molecules ]的添加顺序一定要按照packmol中的顺序添加,否则不匹配。这个是经过离子平衡后的top
其中3个重要的itp如下:
metal.itp
[ atomtypes ]
; type at# mass charge ptype sig eps
MP 6 0.01 0.000 A 3.55000e-01 2.92880e-01
MM 6 0.01 -0.000 A 3.55000e-01 2.92880e-01
mPlus.itp
[ moleculetype ]
;Name nrexcl
MP 1
[ atoms ]
; id at type res nr residu name at name cg nr charge mass
1 MP 1 MP MP 1
mMinus.itp
[ moleculetype ]
;Name nrexcl
MM 1
[ atoms ]
; id at type res nr residu name at name cg nr charge mass
1 MM 1 MM MM 1
离子平衡
gmx grompp -f ions.mdp -c two_level.gro -p two_level.top -o two_level_ions.tpr
gmx genion -s two_level_ions.tpr -o two_level_ions_finish.gro -p two_level.top -pname NA -np 31
然后替换sol
制作索引
gmx make_ndx -f two_level_em.gro -o two_level_em.ndx
能量最小化
gmx grompp -f em.mdp -c two_level_ions_finish.gro -p two_level.top -o two_level_em.tpr
gmx mdrun -v -deffnm two_level_em
em.mdp
; Run Control
integrator = steep ; A steepest descent algorithm for energy minimization
emtol = 100 ; Stop minimization when the maximum force < 100.0 kJ/mol/nm
emstep = 0.01 ; [nm] initial step-size
nsteps = 50000 ; Maximum number of (minimization) steps to perform
; Output Control
nstlog = 1 ; # of steps that else between writing energies to the log file
nstenergy = 1 ; # of steps that else between writing energies to energy file
; Neighbor searching and nonbonded interaction
cutoff-scheme = verlet ; Cutoff-related parameters
nstlist = 1 ; Frequency to update the neighbor list and the long-range forces
ns_type = grid ; Method to determine neighbor list (simple, grid)
pbc = xyz ; Use periodic boundary conditions in all directions
rlist = 1.2 ; [nm] Cut-off for making neighbor list (short range forces)
; Electrostatics
coulombtype = PME ; Treatment of long range electrostatic interactions
rcoulomb = 1.2 ; [nm] Short-range electrostatic cut-off
; Van der waals
vdwtype = cutoff
rvdw = 1.2 ; [nm] Short-range Van der Waals cut-off
; Temperature and pressure coupling are off during EM
tcoupl = no ; No temperature coupling
pcoupl = no ; No pressure coupling (fixed box size)
; No velocities during EM
gen_vel = no ; Do not generate velocities
; options for bonds
constraints = none ; No contraints except those defined in the topology
nvt平衡
gmx grompp -f nvt.mdp -c two_level_em.gro -p two_level.top -o two_level_nvt.tpr
gmx mdrun -v -deffnm two_level_nvt
nvt.mdp
; Run Control
integrator = md ; leap-frog integrator of Newton's equations of motion
tinit = 0 ; [ps] starting time for the run
dt = 0.002 ; 2 fs - [ps] time step for integration
nsteps = 50000 ; 0.1 ns
; Output Control
nstxout = 1000 ; save coordinates every 2 ps
nstenergy = 500 ; save energies every 1 ps
nstlog = 50000 ; update log file every 100 ps
; Bond parameters
continuation = no ; apply constraints to the start configuration and reset shells
constraints = h-bonds ; convert all bonds to constraints
constraint-algorithm = lincs ; holonomic constraints
; Neighbor searching and nonbonded interactions
cutoff-scheme = verlet ; Cutoff-related parameters
nstlist = 20 ; Frequency to update the neighbor list and the long-range forces
ns_type = grid ; Method to determine neighbor list (simple, grid)
pbc = xy ; Use periodic boundary conditions in all directions
rlist = 1.2 ; [nm] Cut-off for making neighbor list (short range forces)
ewald-geometry = 3dc
nwall = 2
wall-atomtype = MW MW
wall-type = 12-6
; Electrostatics
coulombtype = PME ; Treatment of long range electrostatic interactions
rcoulomb = 1.2 ; [nm] Short-range electrostatic cut-off
; Van der waals
vdwtype = cutoff
rvdw = 1.2 ; [nm] Short-range Van der Waals cut-off
; Do I need pme-order and fourier spacing?
;pme_order = 4 ; cubic interpolation
;fourierspacing = 0.16 ; grid spacing for FFT
; Temperature coupling
tcoupl = v-rescale ; Temperature coupling, modified Berendsen thermostat
tc_grps = system ; Group to couple to separate temperature baths
tau_t = 0.1 ; [ps] Time constant for coupling
ref_t = 300 ; [K] Reference temperature for coupling
; Pressure coupling is off for NVT
Pcoupl = No ; No pressure coupling (fixed box size)
; Generate velocities to start
gen_vel = yes ; assign velocities from Maxwell distribution
gen_temp = 300 ; temperature for Maxwell distribution
gen_seed = -1 ; generate a random seed
; Freeze Sheets
freezegrps = MP MM
freezedim = Y Y Y Y Y Y
这个结构没有崩溃,然后计算相关介电常数
gmx dipoles -corr total -c two_level.xvg -f two_level_nvt.trr -s two_level_nvt.tpr
gmx dielectric -f two_level.xvg -eps0 7.87146 -ffn aexp -o two_level_freq.xvg -nsmooth 10
xmgrace two_level_freq.xvg
相关结果如下

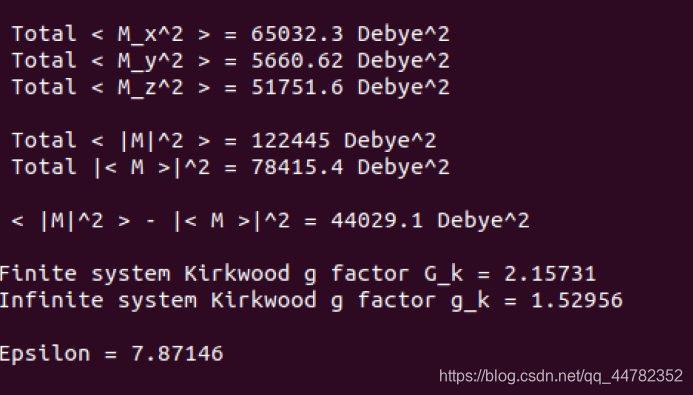
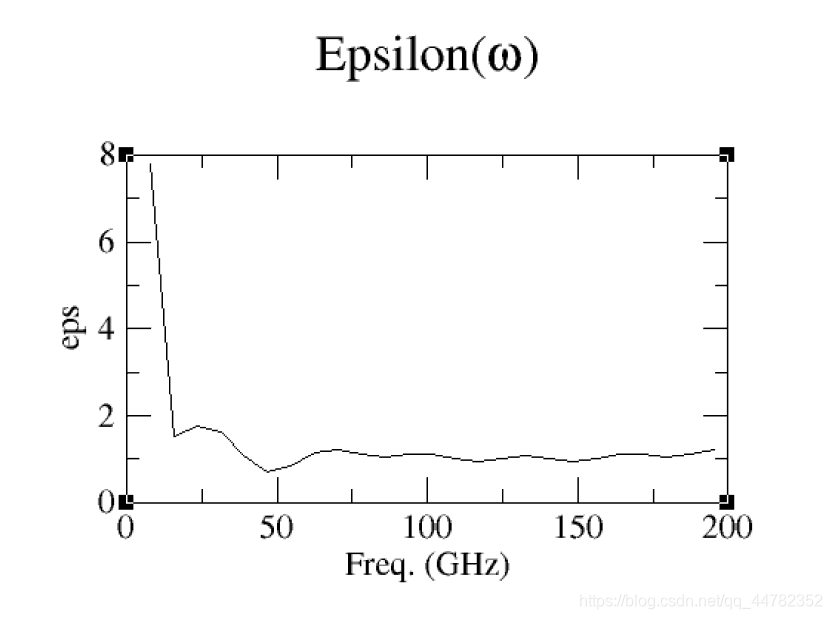
水从边上漏出去吸到蛋白上了,但是整体的结构是呈现电中性的。


















 1526
1526

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











