









小罗碎碎念
今天这期推文是一期总结性推文,系统地回顾了我5月阅读的所有文献。
正所谓温故而知新,在翻阅从前阅读过的文章时,发现自己遗落了不少的内容,又或者说当时的知识储备量,还不足以发现那些内容的重要性。今天这五篇文献是我从几十篇文献中筛选出来的,并且都是与病理组学相关的开源项目。最近一直在整理一些高质量的开源项目,试图从前辈们那里获取一点经验,让自己的项目也变得更加“平易近人”一些。
说实话,复现项目其实是一个很麻烦的过程,因为初学者大概率不知道作者最终呈现给你的代码是哪一个版本——有的项目同时涉及到分类和分割,不同的任务在预处理过程就不同了,还在傻傻的跟着readme文档走(不同项目的readme文档真的一言难尽)。
此外,作者在搭建仓库时随手的一个失误就会让你浪费好几天的时间,这些都是无法避免的。除去这些,如何修改路径、修改config、放置你的数据都会给刚入门的你造成困难。上面说的这些都是我亲身经历后的一些感悟总结,因此才会有了这一篇推文。关于项目复现的推文和教程其实很少,难写更难讲,且看且珍惜,哈哈。

一、数字组织学标志物HiPS:提升侵袭性乳腺癌预后预测的新工具

一作&通讯
| 作者角色 | 作者姓名 | 单位名称(中文) |
|---|---|---|
| 第一作者 | Mohamed Amgad | 西北大学范伯格医学院病理学系,芝加哥,伊利诺伊州,美国 |
| 通讯作者 | Lee A. D. Cooper | 西北大学范伯格医学院病理学系,芝加哥,伊利诺伊州,美国 |
文献概述
这篇文章报道了一种新的数字组织学生物标志物HiPS的开发和验证,该标志物能够通过深度学习分析肿瘤微环境的形态学特征,以提高侵袭性乳腺癌的预后预测准确性。
研究团队由Mohamed Amgad等人组成,他们开发了一种名为HiPS(Histomic Prognostic Signature)的数字组织学生物标志物,用于提高侵袭性乳腺癌预后的效果。
- 乳腺癌是一种全球最常见的恶性肿瘤,具有高度异质性,其生存结果因肿瘤生物学、治疗方案和社会经济因素而有很大差异。
- 传统上,病理学家使用Nottingham标准对乳腺组织的微观外观进行定性分级,但这种方法不能准确反映肿瘤微环境中的非癌性元素。
- HiPS利用深度学习技术,通过映射细胞和组织结构来量化上皮、基质、免疫和空间交互特征,从而提供一种全面的、可解释的肿瘤微环境形态学评分系统。
- HiPS的开发基于美国癌症协会的Cancer Prevention Study-II(CPS-II)中的大规模前瞻性队列,并使用来自三个独立队列的数据进行了验证,包括前列腺、肺、结直肠和卵巢癌试验(PLCO)、癌症预防研究-3(CPS-3)和癌症基因组图谱(TCGA)。
- HiPS在预测生存结果方面一致优于病理学家的预测,且与肿瘤-淋巴结-转移(TNM)分期和相关变量无关。
- 研究还发现,HiPS特征与高基因组不稳定性、低氧免疫微环境、肌成纤维细胞(myCAF)表型和缺乏CD8+ T淋巴细胞的次优免疫反应有关。
研究的结论是,HiPS是一个经过稳健验证的生物标志物,可以支持病理学家的工作并改善患者的预后。这项研究提供了一种新的方法,通过深入分析肿瘤微环境的形态学特征,来提高对乳腺癌患者预后的预测准确性。
代码仓库
-
HistomicsTK - 这是一个用于数字病理图像处理的Python库。在文章中,它被用于提取组织样本的形态学特征,这是HiPS模型开发的基础工作之一。
- 链接:HistomicsTK
- 版本:v.1.2.10
-
histolab - 这是一个Python库,用于数字病理图像的预处理。在研究中,它被用于处理和分析全切片图像(WSIs)。
- 链接:histolab
- 版本:v.0.6.0
-
scikit-image - 这是一个Python库,提供图像处理功能。在文章中,它被用于提取细胞核的形态学特征。
- 链接:scikit-image
- 版本:v.0.18.0
-
Lifelines - 这是一个Python库,用于生存分析。在研究中,它被用于分析患者的生存数据。
- 链接:Lifelines
- 版本:v.0.27.8
-
GSEAPy - 这是一个Python实现的基因集富集分析(Gene Set Enrichment Analysis, GSEA)工具。在研究中,它被用于分析基因表达数据,以探索HiPS特征与基因表达之间的关系。
- 链接:GSEAPy
- 版本:v.1.0.6
-
U-Net - 这是一个用于医学图像分割的卷积神经网络架构。在文章中,它被用作构建MuTILs模型的基础,用于全切片图像的区域和细胞核分割。
- 链接:U-Net
-
HookNet - 这是一个多分辨率卷积神经网络,用于病理组织学全切片图像的语义分割。
- 链接:HookNet
二、自动化三级淋巴结构分析:一种新的癌症预后和免疫治疗反应预测工具

一作&通讯
| 角色 | 姓名 | 单位名称(中文) |
|---|---|---|
| 第一作者 | 陈 (Ziqiang Chen) | 复旦大学基础医学院生物化学与分子医学系 |
| 并列第一作者 | 王 (Xiaobing Wang) | 中国医学科学院肿瘤医院,国家癌症中心,北京协和医学院 |
| 并列第一作者 | 金 (Zelin Jin) | 复旦大学基础医学院生物化学与分子医学系 |
| 并列第一作者 | 李 (Bosen Li) | 复旦大学中山医院普外科/胃癌中心 |
| 通讯作者 | 焦 (Yuchen Jiao) | 中国医学科学院肿瘤医院,国家癌症中心,北京协和医学院 |
| 通讯作者 | 应 (Jianming Ying) | 中国医学科学院肿瘤医院,国家癌症中心,北京协和医学院 |
| 通讯作者 | 刘 (Zhihua Liu) | 复旦大学中山医院普外科/胃癌中心 |
| 通讯作者 | 刘 (Yun Liu) | 复旦大学基础医学院生物化学与分子医学系 |
文献概述
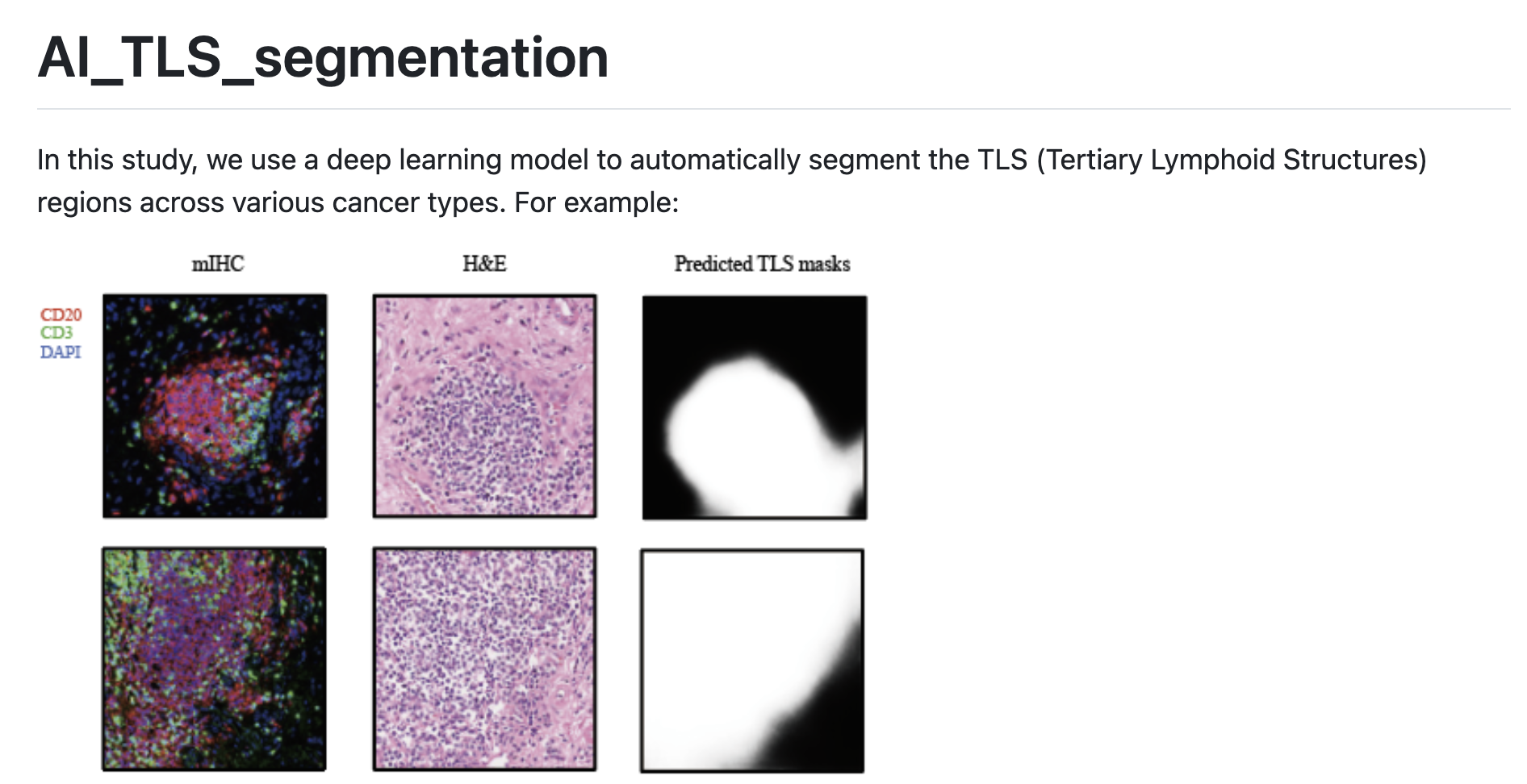
这篇文章报道了一个深度学习模型的开发,该模型能够从常规的H&E染色全切片图像中自动分割和量化三级淋巴结构(Tertiary Lymphoid Structures, TLS),为预测癌症预后和免疫疗法反应提供了一种新的自动化方法。
研究团队开发了一个深度学习模型,自动化地从苏木精-伊红(Hematoxylin and Eosin, H&E)全切片图像(Whole-Slide Images, WSIs)中分割和计算TLS的比率。通过与多项免疫组化(multiplex immunohistochemistry, mIHC)染色的图像数据进行比较,研究证明了模型的准确性和可靠性。
研究的主要发现包括:
- TLS的存在与多种癌症中积极的免疫疗法反应和预后有关。
- 传统的mIHC染色方法虽然可以识别TLS,但资源密集且不普遍可用。
- 深度学习模型在内部测试集上达到了0.91的Dice系数和0.819的交并比(Intersection over Union, IoU),在外部验证集上分别达到了0.866和0.787。
- TLS比率与6140名患者中的B淋巴细胞水平和CXCL13的表达相关,这些患者涵盖了16种肿瘤类型。
- 包括TLS比率的预后模型显著提高了模型的区分能力,特别是在10种不同的肿瘤类型中。
- 在多个独立队列中,较高的TLS比率预测了免疫疗法的积极反应。
研究的结论是,基于深度学习的TLS分割和量化方法为预测免疫疗法反应和癌症预后提供了一种自动化和可重复的方法,突出了其在精准肿瘤学中的潜力。
代码仓库
https://github.com/zonechen1994/AI_TLS_segmentation
三、人工智能在胎盘病理学中的应用

一作&通讯
| 角色 | 姓名 | 单位名称(英文) | 单位名称(中文) |
|---|---|---|---|
| 第一作者 | Claudia Vanea | Nuffield Department of Women’s & Reproductive Health, University of Oxford, UK | 牛津大学纳菲尔德妇幼保健系 |
| 通讯作者1 | Cecilia M. Lindgren | Big Data Institute, Li Ka Shing Centre for Health Information and Discovery, University of Oxford, UK | 牛津大学李嘉誠健康资讯与发现大数据研究所 |
| 通讯作者2 | Christoffer Nellåker | Nuffield Department of Women’s & Reproductive Health, University of Oxford, UK | 牛津大学纳菲尔德妇幼保健系 |
文献概述
这篇文章介绍了一种名为HAPPY(Histology Analysis Pipeline.PY)的深度学习工具,它能够高精度地分析人类胎盘组织学全切片图像,量化细胞和组织结构,为胎盘健康评估提供了新的量化指标。
HAPPY方法与传统的基于图像块的特征或分割方法不同,它遵循可解释的生物学层次结构,能够在全切片图像中以单细胞分辨率表示组织内的细胞和细胞群。研究人员使用HAPPY对健康足月胎盘的细胞和组织结构进行了量化分析,并将其作为评估胎盘健康的基础指标。此外,他们还展示了这些指标在具有临床意义的胎盘梗死中的偏差。
HAPPY通过三个阶段的深度学习流程来实现:
- (i) 核定位的物体检测模型
- (ii) 细胞分类的图像分类模型
- (iii) 用于组织分类的图神经网络(GNN)
该方法在11,755个核和13,842个细胞上训练和验证核定位和细胞分类模型,并在独立的测试集上进行评估。此外,还在468,869个节点上训练和验证图神经网络组织分类模型,并在179,095个测试节点上进行评估。
研究人员比较了HAPPY的组织分类模型与四位实践围产期病理学家的标签和Cohen’s kappa一致性得分,以验证训练注释和模型性能。结果表明,HAPPY的细胞和组织预测与独立临床专家的预测和胎盘生物学文献紧密匹配。
此外,文章还讨论了HAPPY方法的泛化能力和领域偏移问题,即模型对不同染色强度的H&E染色的不变性。通过数据增强,包括定制的H&E染色增强,研究了核检测和细胞分类模型对染色不变性的影响。研究结果表明,使用数据增强训练的模型在不同机构的数据上具有更好的泛化能力。
最后,文章讨论了HAPPY方法的潜在应用,包括作为围产期病理学家的数字组织病理学工具,以及在胎盘研究中推动对人类胎盘及其机制的理解。作者还提到了HAPPY方法的潜在改进,例如提高地面真实注释的质量,扩展细胞分析以包括其他细胞类型,并使用转移学习将当前的胎盘模型作为领域相关预训练基础应用于其他器官组织学。
代码仓库
-
HAPPY的代码库链接:
- 原文引用:“The HAPPY codebase, training data, and trained models for placenta histology are available at (https://github.com/Nellaker-group/happy).”
- 作用:提供了HAPPY(Histology Analysis Pipeline.PY)的代码库,其中包括用于胎盘组织学全切片图像分析的深度学习模型、训练数据和训练好的模型,供研究者下载和使用。
-
GTEx数据集链接:
- 原文引用:“To show that this is valid in principle, we have additionally conducted a preliminary investigation of our nuclei localisation and cell classification models across WSIs of a placenta membrane roll, umbilical cord, a second-trimester placenta with chorioamnionitis, and also across WSIs of other organs in the GTEx dataset (Supplementary Fig. 1).”
- 作用:提到了GTEx(Genotype-Tissue Expression)数据集,这是一个公开的基因型-组织表达项目数据集,用于支持文中提及的模型在不同组织上的初步研究。
-
数据和代码的Zenodo存档链接:
- 原文引用:“The datasets generated for training and validating each deep learning model along with trained model weights are available for download at the Google Drive link: https://tinyurl.com/happy or from Zenodo: 10.5281/zenodo.10535021 with no restrictions.”
- 作用:提供了一个DOI链接到Zenodo,这是一个开放获取的数据存储库,用于存储和分享研究数据和代码,确保研究的可重复性和透明度。
-
HAPPY的GitHub链接:
- 原文引用:“Code is available at the following GitHub repository https://github.com/Nellaker-group/happy and at https://doi.org/10.5281/zenodo.1052923996.”
- 作用:提供了HAPPY项目的GitHub仓库链接,研究者可以从这里获取源代码、训练模型和相关文档,以便进一步的研究和应用。
四、高多重免疫荧光成像技术HIFI:洞察脑肿瘤微环境的空间重组

一作&通讯
| 角色 | 姓名 | 单位(中文) |
|---|---|---|
| 第一作者 | Spencer S. Watson | 洛桑大学肿瘤系、洛桑大学路德维希癌症研究所、洛桑Agora癌症研究中心、洛桑大学医院神经肿瘤研究中心 |
| 通讯作者 | Johanna A. Joyce | 同上,以及剑桥大学机器智能实验室、英国癌症研究大挑战iMAXT联盟 |
文献概述
这篇文章报道了一种新的高多重标记成像技术HIFI,用于揭示脑肿瘤经放疗和复发后肿瘤微环境的空间重组织。
研究主要关注了肿瘤微环境(TME)在脑肿瘤放疗和复发后的变化,并介绍了一种名为Hyperplexed Immunofluorescence Imaging (HIFI) 的空间蛋白质组学工作流程。
-
研究背景:肿瘤微环境在治疗反应中扮演关键角色,涉及细胞景观、空间组织和细胞外基质组成的相互联系的变化。然而,由于现有高维成像技术的局限性和肿瘤内部异质性的广泛性,同时从空间角度评估这些变化具有挑战性。
-
HIFI技术介绍:HIFI技术克服了现有方法的局限,允许在高倍镜下同时分析超过45种标记物,并使用成本效益高的高通量工作流程。
-
研究方法:研究者将HIFI与机器学习特征检测、基于图的网络分析和基于聚类的邻域分析相结合,分析了胶质母细胞瘤的前临床模型和乳腺癌转移到大脑的模型对放疗的微环境反应,并比较了这两种模型的反应差异。
-
研究发现:研究表明,胶质母细胞瘤在放疗后会经历免疫细胞群体和结构架构的广泛空间重组织,而脑转移瘤则没有显示出类似的重组织。这种综合的空间分析揭示了不同脑肿瘤模型对放疗反应的高度差异性。
-
胶质母细胞瘤的治疗:胶质母细胞瘤是成人最常见的也是最具侵袭性的原发性脑肿瘤,尽管采用了手术切除、电离辐射和替莫唑胺基础化疗等标准治疗,但中位生存期仅为14个月,5年生存率不到5%。
-
HIFI工作流程:HIFI工作流程设计简单,使用开源软件和常规实验室设备即可实施。它包括循环免疫荧光成像、全切片图像对齐和配准、机器学习结构注释、深度学习细胞分割和基于聚类的细胞分类。
-
实验样本:使用了RCAS-hPDGF-B; Nestin-Tv-a; Ink4a/Arf KO基因工程小鼠模型来模拟胶质母细胞瘤,并与乳腺癌转移到大脑的模型进行了比较。
-
图像分析:通过深度学习算法StarDist进行细胞分割,并通过FlowSOM进行半监督细胞分类,以识别不同的细胞类型。
-
研究意义:HIFI方法是非破坏性的,允许大规模成像,并且适用于高通量分析。它是低成本和开源的,可以被广泛地采用和访问。
-
结论:研究提供了一种新的策略,用于在肿瘤发展过程中以及治疗干预后研究TME,使研究人员能够深入和全面地审视多细胞区域相互作用。
文章强调了HIFI技术在癌症研究中的应用潜力,尤其是在理解肿瘤微环境如何响应治疗并影响肿瘤复发方面。
代码仓库
这篇文章中提到了两个代码链接,它们在文章中的作用如下:
-
HIFI Alignment Tool:
- 链接: https://github.com/jhausserlab/HiFiAlignmentTool
- 作用: 这是一个用于HIFI(Hyperplexed Immunofluorescence Imaging)图像数据对齐的工具。在HIFI技术中,需要将多个成像周期的图像数据重新组合成全分辨率的图像。这个工具帮助研究人员将不同成像周期的DAPI通道对齐,并将变换应用于每个图像通道,最终将对齐的图像保存为单一的OME TIFF文件,为后续的细胞分割和分析提供基础。
-
StarDist cell segmentation model and associated training data:
- 链接: https://github.com/TristanWhitmarsh/IMAXT_StarDist_Cellpose
- 作用: StarDist是一个基于深度学习的细胞分割算法,用于从复杂的生物医学图像中准确检测和分割细胞。在这项研究中,StarDist被用来处理HIFI图像数据,通过训练数据集生成模型,从而实现对细胞核的精确分割。这对于理解肿瘤微环境中的细胞组成和空间关系至关重要。
五、基于回归的深度学习:从病理切片预测分子生物标志物

一作&通讯
| 角色 | 姓名 | 单位(中文) |
|---|---|---|
| 第一作者 | Omar S. M. El Nahhas | 德累斯顿工业大学医学系(TUD Dresden University of Technology, Dresden, Germany) |
| 通讯作者 | Jakob Nikolas Kather | 德累斯顿工业大学医学系(TUD Dresden University of Technology, Dresden, Germany) |
文献概述
这篇文章报道了一种基于回归的深度学习方法,能够从病理切片图像中准确预测连续的分子生物标志物,为计算病理学和精准医疗提供了新的工具。
这项研究由Omar S. M. El Nahhas和Jakob Nikolas Kather带领的团队进行,他们开发并评估了一种自监督的、基于注意力机制的弱监督回归方法,用于直接从11,671张涵盖九种癌症类型的患者的图像中预测连续的生物标志物。研究发现,使用回归方法可以显著提高生物标志物预测的准确性,并且与分类方法相比,能够更好地对应已知临床相关区域的预测。
文章提到,传统的深度学习方法通常将生物标志物预测限制在分类问题上,但许多生物标志物实际上是连续的测量值。研究团队提出假设,回归方法在弱监督分析中的表现可能优于分类方法。为了验证这一点,他们开发了一种名为CAMIL(contrastively-clustered attention-based multiple instance learning)的回归方法,结合了自监督学习和注意力机制,并与现有的分类方法和另一种回归方法进行了比较。
研究结果显示,CAMIL回归方法在预测同源重组缺陷(HRD)评分等多个临床和生物学上相关的生物标志物方面,具有更高的准确性和预测价值。此外,CAMIL回归方法在预测肿瘤微环境中的关键生物过程标志物方面,也显示出比分类方法更好的性能。
这项研究还探讨了回归方法在预测已知临床相关区域的生物标志物方面的有效性,并通过注意力热图进行了可视化分析。结果表明,CAMIL回归方法生成的注意力热图在81%的情况下比分类方法更准确地对应已知临床相关区域。
最后,研究还评估了CAMIL回归方法在预测结直肠癌患者生存方面的能力。在DACHS研究的2297名结直肠癌患者的大型队列中,回归方法预测的生物标志物在生存预测方面显示出比分类方法更高的预后价值。
文章强调,CAMIL回归方法作为一种开源工具,为计算病理学中的连续生物标志物分析提供了一个有希望的替代方案,并可能对精准医疗领域产生重要影响。
代码仓库
-
预处理管道:
- 链接:https://github.com/KatherLab/end2end-WSI-preprocessing/releases/tag/v1.0.0-preprocessing
- 作用:这个链接提供了用于处理整个幻灯片图像(WSI)的预处理管道,它包括将WSI切割成小块、去模糊、背景去除等步骤,为后续的图像分析和模型训练做准备。
-
分类管道:
- 链接:https://github.com/KatherLab/marugoto/releases/tag/v1.0.0-classification
- 作用:这个链接包含了用于分类任务的深度学习模型管道,它基于注意力机制的多重实例学习(attMIL)方法,用于分类预测病理切片图像中的生物标志物。
-
回归管道:
- 链接:https://github.com/KatherLab/marugoto/releases/tag/v1.0.0-regression
- 作用:这个链接提供了回归模型的管道,它用于预测连续的生物标志物值,与分类方法相比,回归方法可以提供更细致的预测结果,有助于更精确地理解病理切片中的生物标志物。
-
分类和回归注意力热图:
- 链接:https://github.com/KatherLab/highres-WSI-heatmaps/releases/tag/v1.0.0-heatmaps
- 作用:这个链接提供了生成注意力热图的工具,这些热图可以可视化模型在做出预测时关注的病理切片区域,有助于理解模型的决策过程并提高模型的可解释性。
这些代码链接为研究人员提供了必要的工具和框架,以便他们可以复现文章中的研究结果,或者将这些方法应用到自己的研究中。
















 808
808

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









