







小罗碎碎念
这篇发表于Nature Biomedical Engineering的论文,聚焦医学AI在乳腺癌研究中的应用,提出了一种名为ST-Net的深度学习算法,用于整合空间基因表达和乳腺肿瘤形态学信息。

| 作者类型 | 姓名 | 单位 |
|---|---|---|
| 第一作者 | Bryan He | 美国加利福尼亚州斯坦福大学计算机科学系 |
| 通讯作者 | Jonas Maaskola | 瑞典斯德哥尔摩皇家理工学院生物技术学院 |
| 通讯作者 | Joakim Lundeberg | 瑞典斯德哥尔摩皇家理工学院生物技术学院 |
| 通讯作者 | James Zou | 美国加利福尼亚州斯坦福大学计算机科学系;美国加利福尼亚州斯坦福大学电气工程系;美国加利福尼亚州斯坦福大学生物医学数据科学系;美国加利福尼亚州旧金山市Chan-Zuckerberg Biohub |
在研究中,团队利用23名乳腺癌患者的30,612个空间转录组数据和对应的组织病理学图像,训练ST-Net算法。
该算法能够从苏木精 - 伊红染色的图像中,以100μm的分辨率预测102个基因的表达,包括多个乳腺癌生物标志物,且在外部数据集上表现出色。与传统方法相比,ST-Net在捕捉肿瘤内异质性和复杂图像信号方面优势显著。
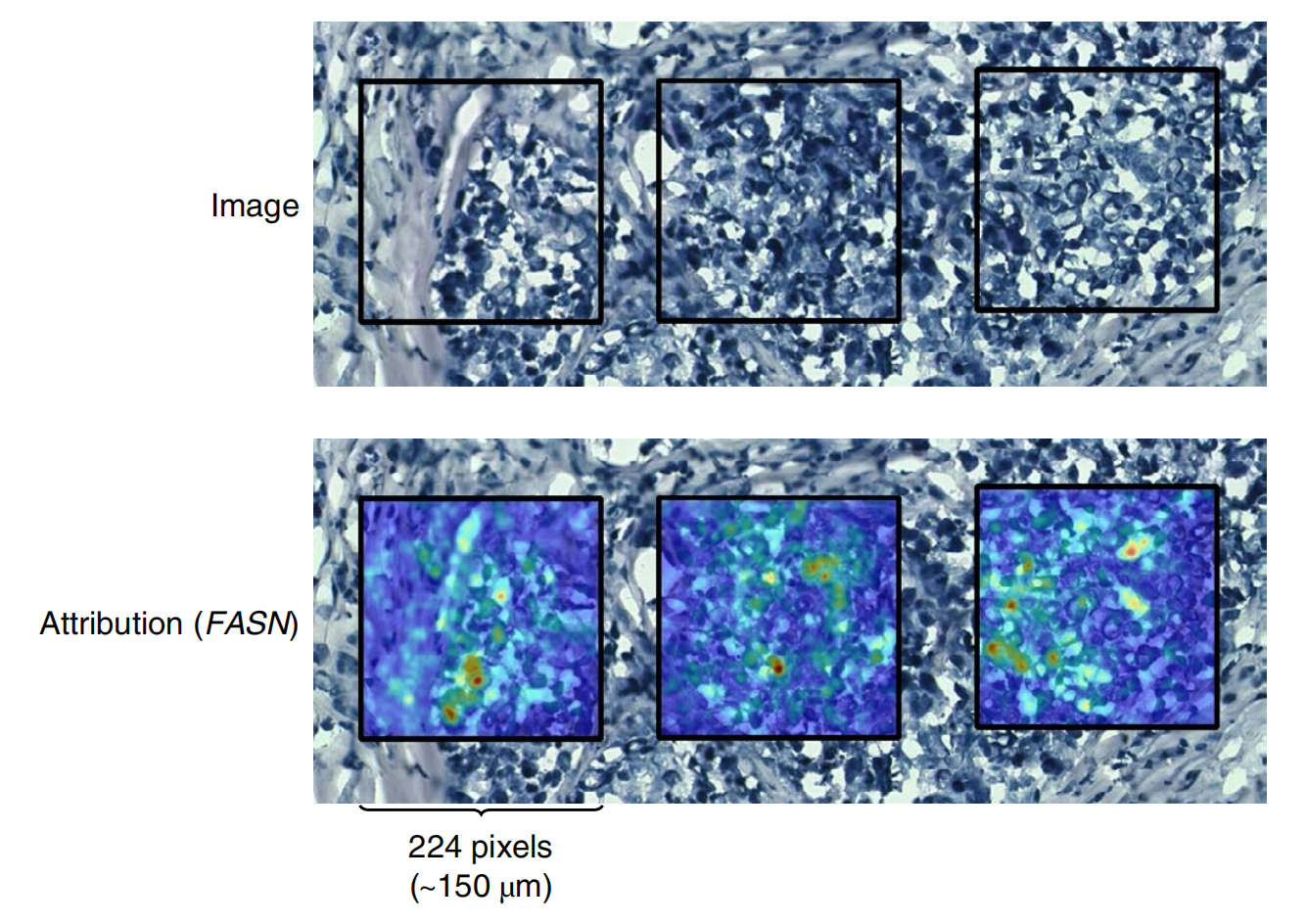
这一研究成果为医学AI领域提供了新的思路和方法,不仅有助于深入理解肿瘤的生物学特性,还为基于图像的分子生物标志物筛查提供了可能。
从事医学AI研究的人员可借鉴此方法,将其拓展应用到其他疾病的研究中,推动医学AI技术在精准医疗领域的进一步发展。
知识星球
如需获取推文中提及的各种资料,欢迎加入我的知识星球!
一、文献概述
“Integrating spatial gene expression and breast tumour morphology via deep learning”发表于Nature Biomedical Engineering,通过开发深度学习算法ST-Net,结合空间转录组学和组织学图像,从苏木精 - 伊红染色的组织病理学图像预测局部基因表达,为研究肿瘤异质性和生物标志物提供了新方法。
- 研究背景:基因表达的空间组织和异质性对组织特性有重要影响,但传统转录组分析技术无法捕获高分辨率空间异质性。空间转录组学可通过DNA条形码区分组织中的不同点,结合组织病理学图像,能获取基因表达的空间信息,但如何整合二者信息成为关键问题。
- 研究方法:开发ST-Net算法,结合空间转录组学和组织学图像,利用卷积神经网络(CNN)从H&E染色的组织病理学图像预测局部基因表达。该算法在来自23名乳腺癌患者的68个乳腺组织切片的30,612个点的新空间转录组学数据集上进行训练。
- 实验结果
- 预测性能良好:ST-Net可在约100μm的分辨率下预测102个基因的空间表达变化,包括多个乳腺癌生物标志物,且在外部数据集(10x Genomics乳腺癌数据和TCGA数据)上表现良好,能可靠地推广到新数据。
- 分析基因通路:通过DAVID进行通路分析,发现102个可预测基因中许多与癌症、药物基因组学和免疫通路相关,表明ST-Net的预测能力在与乳腺癌相关的基因中富集。
- 量化共定位:利用ST-Net量化肿瘤和免疫表达的空间共定位,发现肿瘤和免疫基因共定位频率低于随机预期,这一结果与之前乳腺癌患者肿瘤浸润淋巴细胞低比例的发现一致。
- 检测肿瘤内变异:ST-Net能检测肿瘤内和正常组织内的基因表达变异,可识别潜在的高分辨率生物标志物,其在肿瘤组织和正常组织中的预测结果显示,能捕捉到部分已知癌症生物标志物的表达变化。
- 性能优势明显:与使用病理学家标签、手工制作特征和基于细胞类型组成的预测方法相比,ST-Net能捕获更细粒度的信息、更复杂的图像信号以及组织中细胞类型组成之外的信息,在预测基因表达方面具有更高的准确性。
- 可解释性分析:利用深度学习解释方法,发现ST-Net能将肿瘤相关的视觉特征(如细胞核增大)与高FASN表达相关联;其潜在空间可自然分离肿瘤和正常组织,且能反映斑点之间的语义相似性。
- 研究展望:ST-Net技术框架可应用于其他组织类型,随着更多空间表达数据集的产生和样本制备的标准化,其性能有望进一步提高。未来可将空间转录组学数据与单细胞RNA测序数据整合,以深入研究细胞类型和基因表达的关系。
二、重点关注
2-1:ST-Net的流程和数据
ST-Net(Spatial Transcriptomics - Net)的预测流程
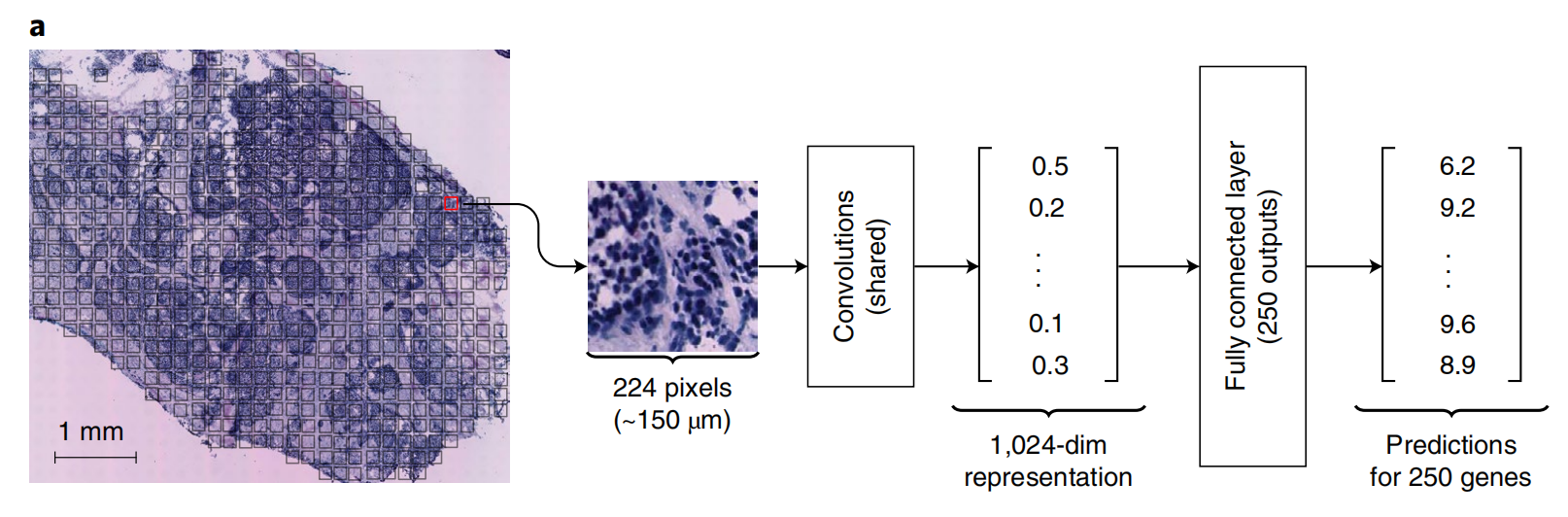
这张图展示了ST-Net(Spatial Transcriptomics - Net)的预测流程:
- 首先,从全切片组织病理学图像中,以空间转录组学的点为中心,提取224×224像素(约150μm)的图像块。
- 接着,这些图像块进入一组共享的卷积层(Convolutions)进行特征提取,得到一个1024维的表示。
- 最后,通过一个有250个输出的全连接层(Fully connected layer),对250个基因进行预测。 该流程用于从组织病理学图像预测基因表达情况。
2-2:采样点数量
患者1有两个切片,其他患者有三个切片,不同患者的采样点数量有所不同 。
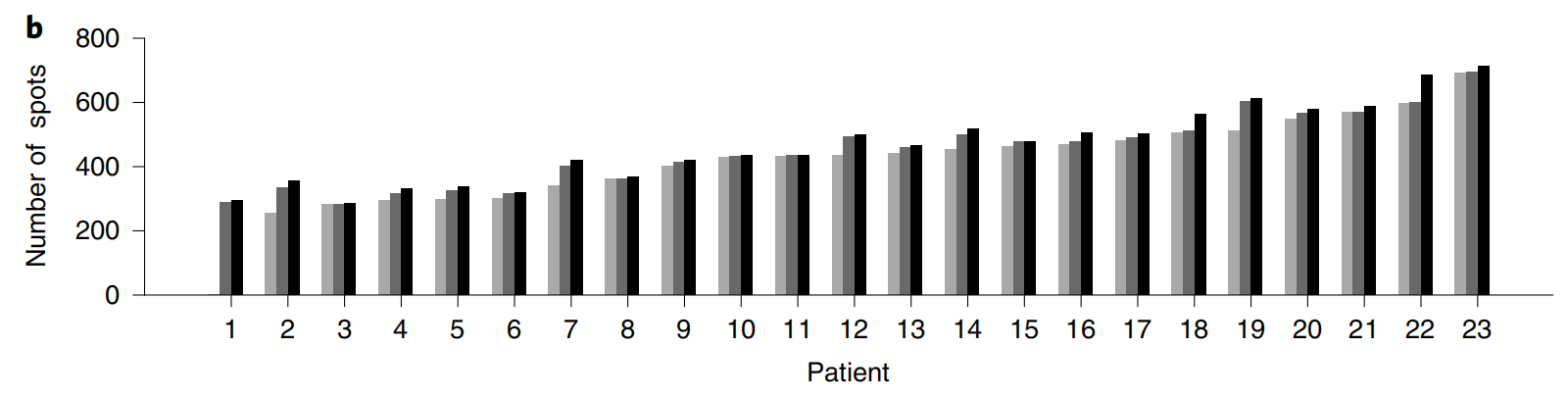
可以看出,患者1和2的采样点数量相对较少,从患者3开始,采样点数量整体呈上升趋势,患者22和23的采样点数量在所有患者中最多。 该图有助于了解不同患者样本的空间转录组学数据的采样规模情况。
2-3:ST - Net预测结果
肿瘤生物标志物FASN
从左到右每列依次为组织病理学图像(Image)、肿瘤区域标注(Tumour,黑点代表肿瘤区域,白点代表正常区域)、FASN基因真实表达情况(True FASN expression)以及ST-Net算法的预测表达(Prediction)。
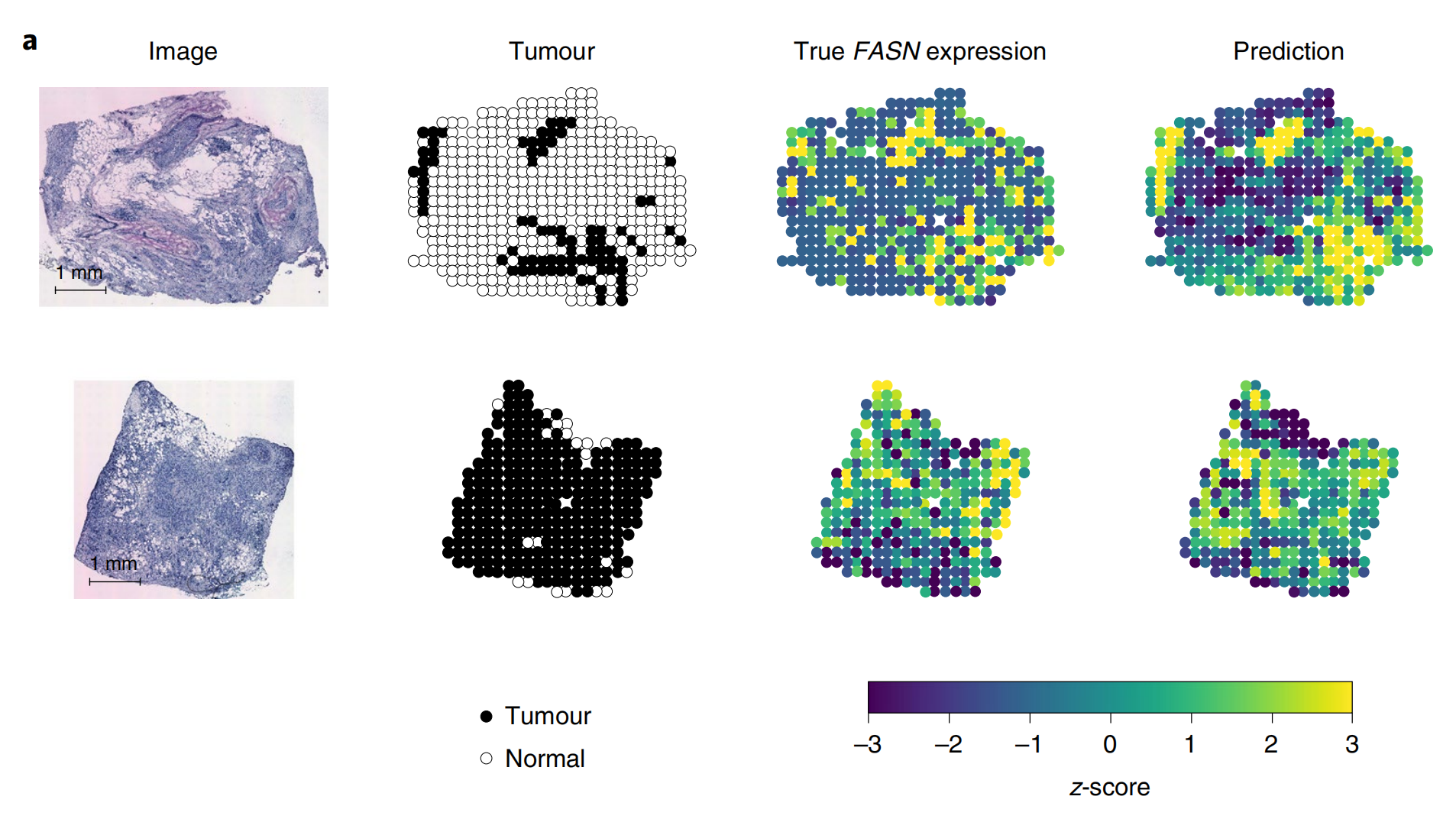
下方的色条表示z分数,用于衡量基因表达水平,不同颜色对应不同的表达强度。通过对比真实表达和预测表达,可评估ST-Net算法在预测FASN基因表达上的效果 。
2-4:ST-Net对前五基因的预测性能
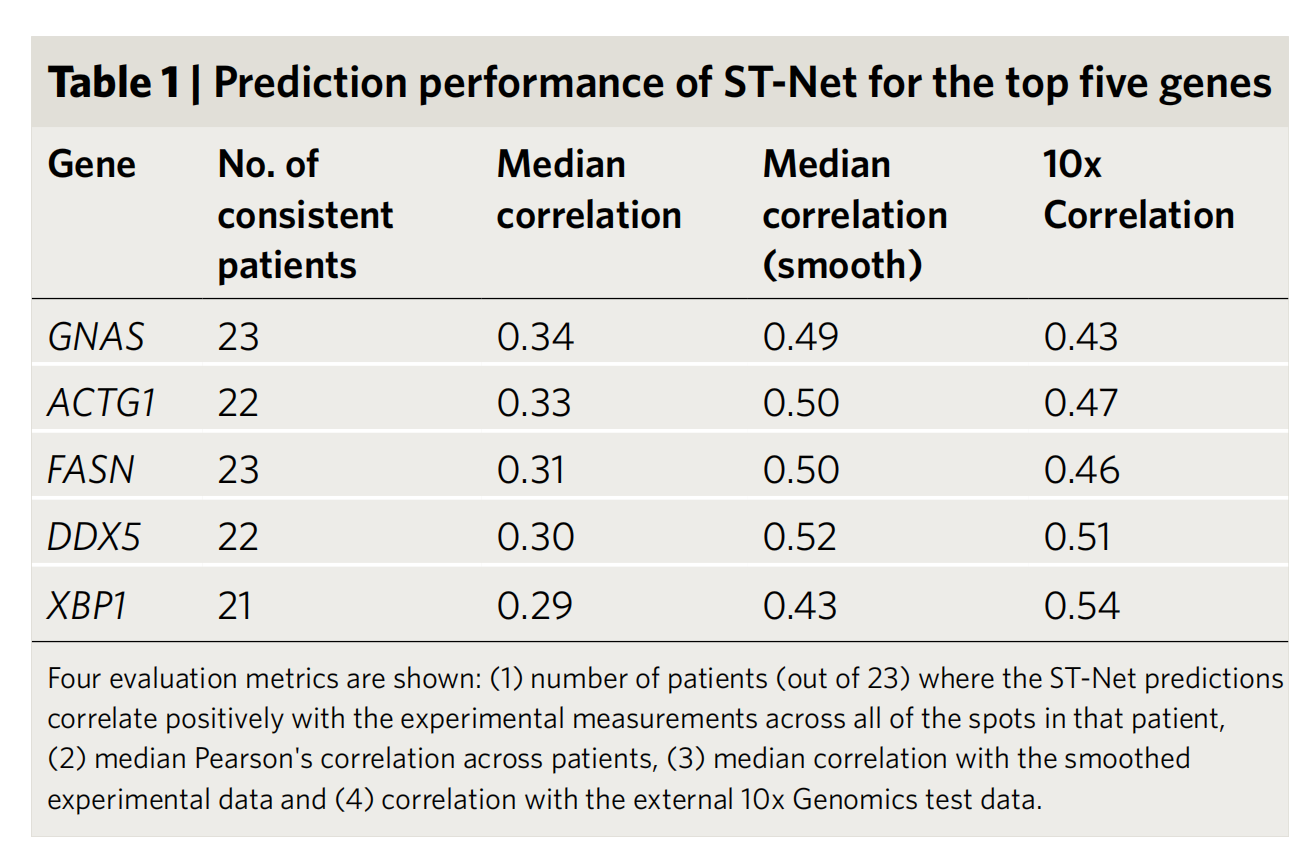
- 指标说明:有四个评估指标,分别是ST-Net预测与实验测量呈正相关的患者数量(共23名患者)、患者间的中位皮尔逊相关系数、与平滑后实验数据的中位相关系数、与外部10x Genomics测试数据的相关系数。
- 结果:
- GNAS基因在23名患者中预测与实验测量正相关,患者间中位相关系数0.34,与平滑数据中位相关系数0.49,与10x数据相关系数0.43。
- ACTG1基因在22名患者中呈正相关,患者间中位相关系数0.33,与平滑数据中位相关系数0.50,与10x数据相关系数0.47。
- FASN基因在23名患者中呈正相关,患者间中位相关系数0.31,与平滑数据中位相关系数0.50,与10x数据相关系数0.46。
- DDX5基因在22名患者中呈正相关,患者间中位相关系数0.30,与平滑数据中位相关系数0.52,与10x数据相关系数0.51。
- XBP1基因在21名患者中呈正相关,患者间中位相关系数0.29,与平滑数据中位相关系数0.43,与10x数据相关系数0.54 。 总体显示ST-Net对这些基因有一定预测能力。
2-5:ST-Net潜在空间的UMAP可视化
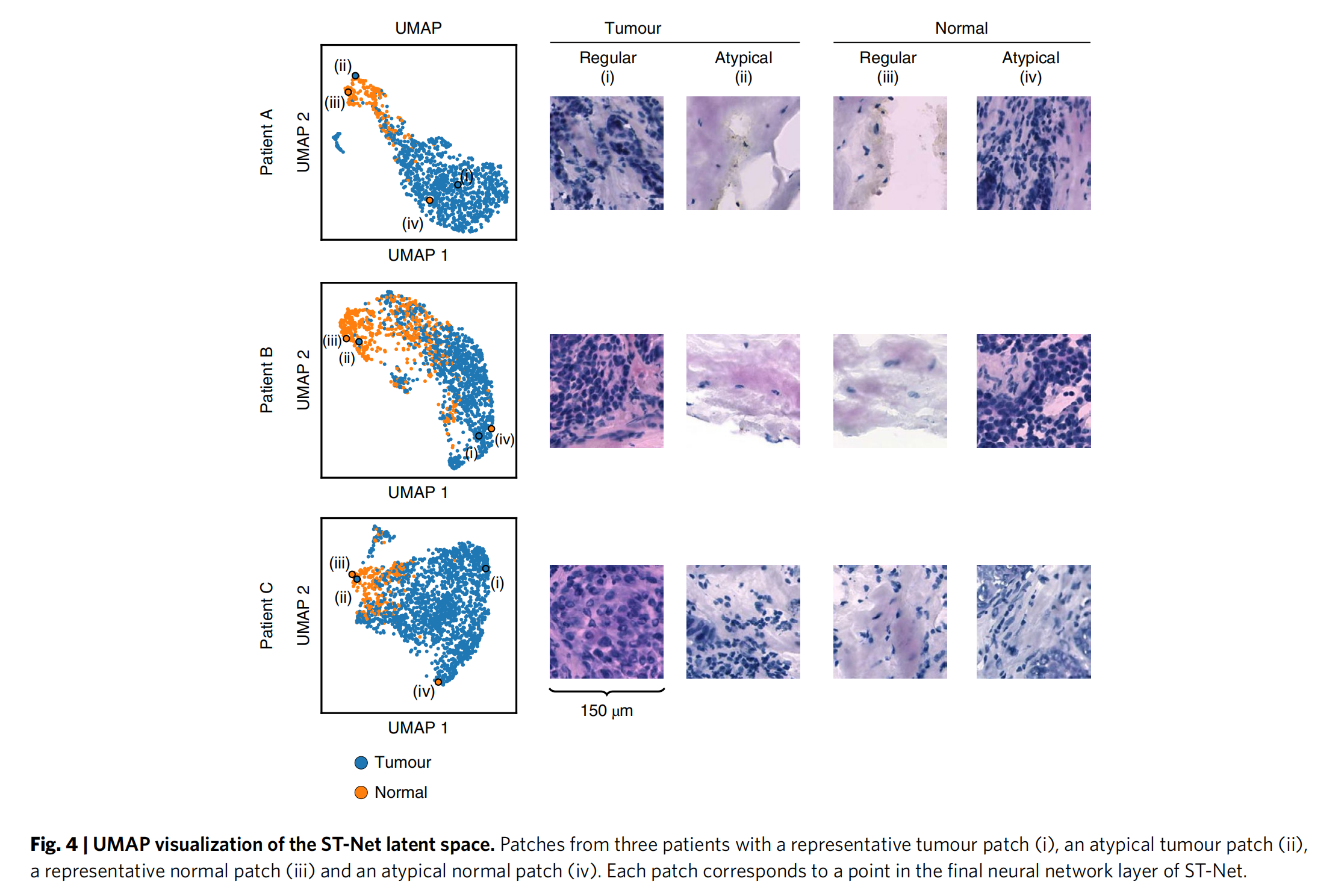
左侧
展示了三位患者(Patient A、Patient B、Patient C)的数据点分布,蓝色代表肿瘤区域,橙色代表正常区域。
可以看出肿瘤和正常区域的数据点在潜在空间中有一定的分离趋势。
右侧
呈现了对应患者的组织病理学图像块,分别为代表性肿瘤图像块(i)、非典型肿瘤图像块(ii)、代表性正常图像块(iii)和非典型正常图像块(iv)。
每个图像块对应ST-Net最终神经网络层中的一个点,通过可视化可以直观地观察到不同组织形态在潜在空间中的分布与特征 。
这有助于理解ST-Net对肿瘤和正常组织的区分能力和潜在空间表示。
三、方法探索
3-1:空间转录组学技术
本研究采用空间转录组学技术(Spatial Transcriptomics)捕获乳腺癌组织切片中mRNA序列的空间分布信息[2]。
通过DNA条形码探针对冷冻组织切片中的mRNA进行空间定位标记,构建测序文库并解析RNA序列的原始空间位置(详见补充图10)。
实验纳入23例乳腺癌患者样本,每例样本包含3张H&E染色组织切片显微图像及对应空间转录组数据(图1a)。空间转录组数据以直径100 µm、中心间距200 µm的网格化斑点(spots)为单位采集RNA表达谱,每例样本斑点数为256-712(图1b),单个斑点可检测数千种mRNA(图1d)。
全数据集共识别26,949种mRNA,每个斑点数据表征为26,949维非负整数向量,反映基因表达计数。
3-2:样本处理与伦理规范
实验遵循Lund大学医院伦理委员会审批的规范[32],患者知情同意后收集乳腺癌活检样本。
样本经快速冷冻、OCT包埋后制备16 µm厚切片,进行H&E染色及标准化成像。
组织区域经病理学注释,包括浸润性癌、原位癌、脂肪组织、纤维组织及免疫细胞区域。
条形码文库的RNA测序与分析均采用标准流程[2,32]。
3-3:ST-Net模型构建与训练
- 模型架构选择:基于ImageNet预训练的DenseNet-121卷积神经网络(CNN)[10-13,33,34],其包含120个卷积层及全连接层。选择该架构主因其参数效率高,适配有限训练数据。
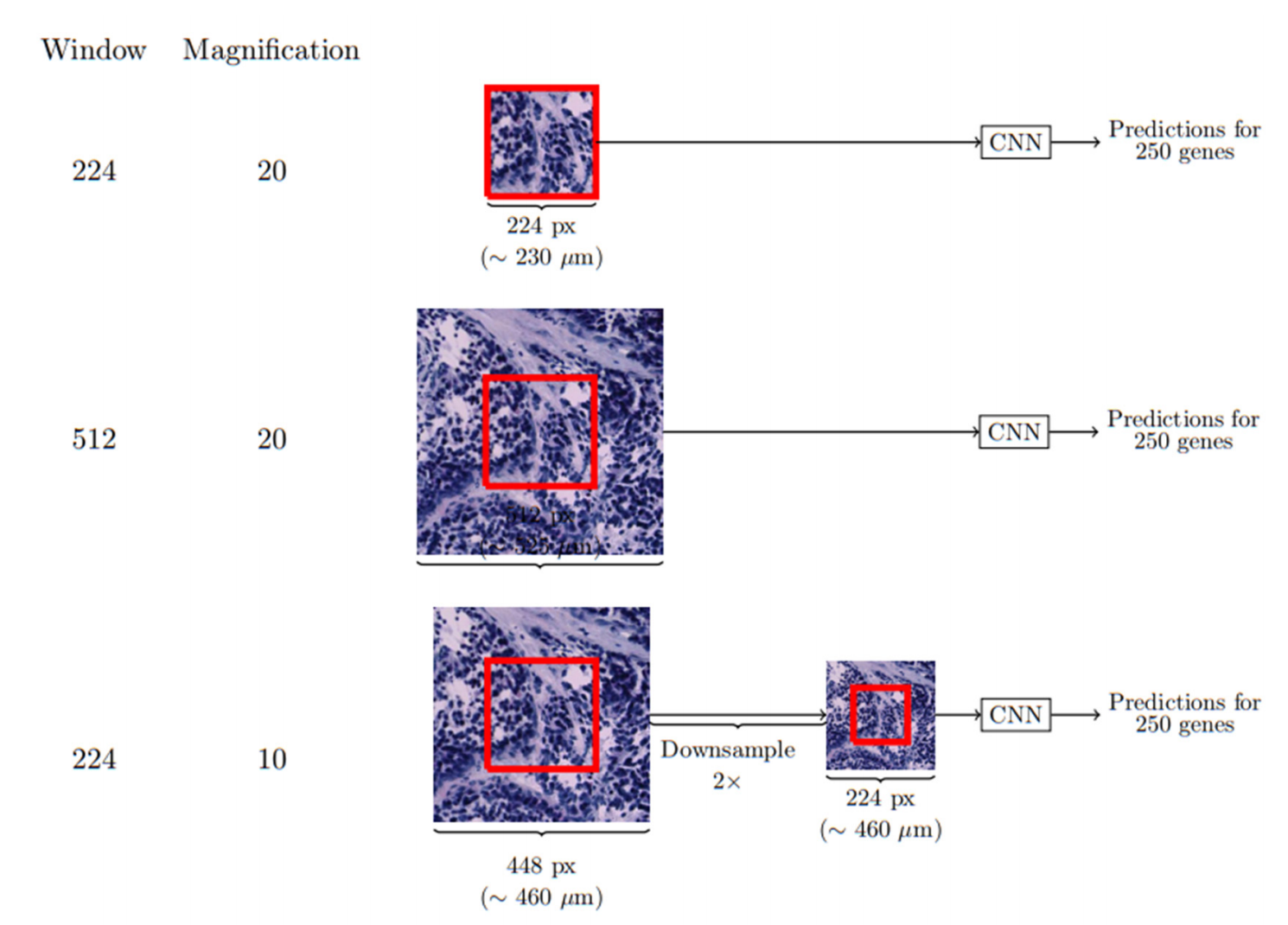
- 输入数据预处理:从全切片图像(约10,000×10,000像素)提取224×224像素的斑块(patch),与空间转录组斑点对应(图1a)。
- 基因表达预测:针对高平均表达的250个基因构建多任务回归模型,共享卷积层权重以提升计算效率,全连接层独立权重用于基因特异性预测。
- 数据标准化:
• 伪计数加1后标准化每斑点总表达量,消除细胞密度对形态学分析的干扰;
• 对标准化计数取对数,缓解基因表达偏态分布(补充图1a)。 - 训练策略:
• 采用留一法交叉验证(Leave-one-out CV),以学习率10⁻⁶、动量0.9的随机梯度下降法优化均方误差;
• 数据增强包括随机旋转(0°、90°、180°、270°)及镜像翻转(50%概率);
• 测试时通过八种对称变换结果平均降低噪声。 - 噪声抑制:通过相邻斑点(3×3邻域)表达量平滑处理模拟深度测序效应。
关键建模决策验证
- 斑块窗口尺寸与放大倍率影响模型精度(补充图11、表4-5);
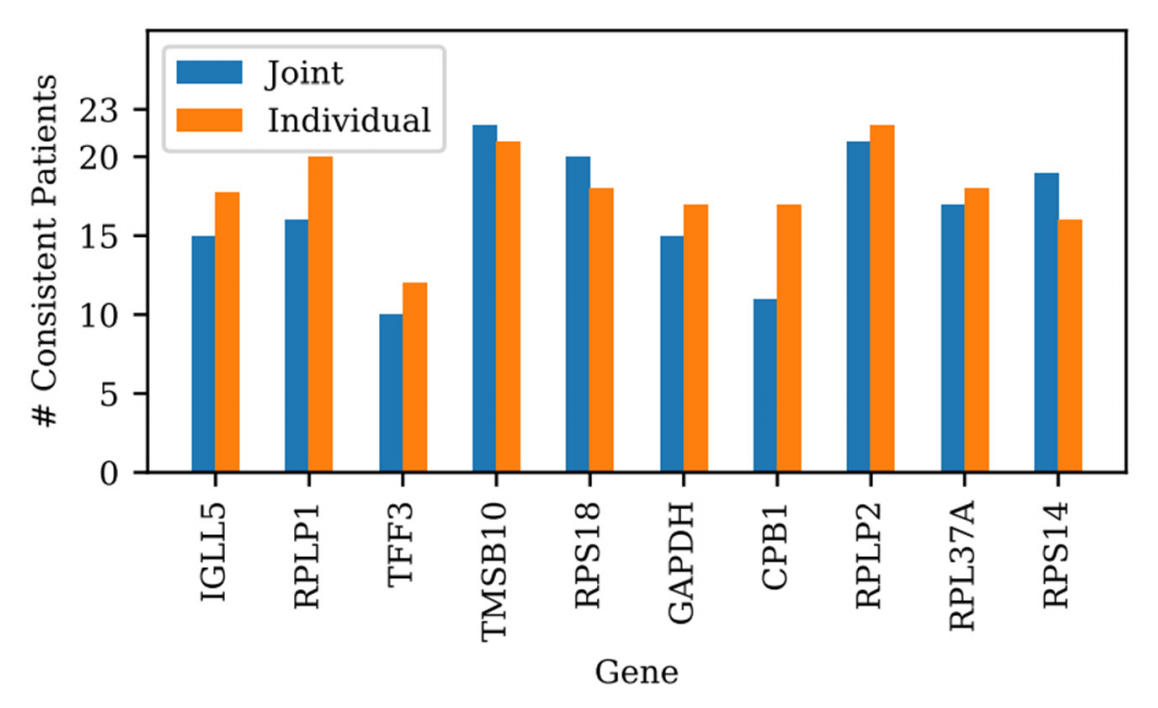
- 基因间权重共享与独立训练性能相近,但共享策略显著降低计算成本(补充图12);
- ImageNet预训练权重可大幅提升模型性能。
3-4:模型比较分析
基于细胞类型组成的模型
- 非负矩阵分解(NMF)模型:通过设定10个因子(对应细胞类型)解析组织斑块(patch)的细胞组成,以交替最小化法迭代200次优化Frobenius范数重构误差(补充图7c显示模型可解释大部分方差)[2]。
- DenseNet-121预测细胞组成:采用与ST-Net相同的超参数与流程训练模型,预测斑块细胞类型组成,并通过NMF模型将细胞组成映射至基因表达谱。
10x Genomics乳腺癌数据分析
基于Visium空间基因表达协议生成的10x Genomics乳腺癌空间转录组数据,应用未经调整的ST-Net模型直接从组织学图像预测基因空间表达水平,并与实验测量值对比[32]。
数据标准化方法与原乳腺癌数据集一致,验证模型泛化能力。
TCGA泛化验证
- 数据预处理:
• 筛选TCGA中1,093例乳腺癌冷冻样本的1,550张全切片图像(×20倍放大),提取512×512像素非重叠组织斑块;
• 通过RGB均值阈值(≤220判定为背景)去除背景占比超50%的斑块。 - 基因表达预测:
• 直接应用原数据集训练的ST-Net模型权重预测TCGA样本基因表达;
• 对比伪批量预测均值与TCGA批量RNA-seq数据,计算基因间Pearson相关性(重点关注肿瘤相关基因AEBP1、BGN、P4HB及免疫相关基因PABPC1、TAGLN2、HLA-B)。 - 亚型分类验证:
• 将TCGA患者按7:3划分为训练集与测试集;
• 基于55个显著正相关基因的伪批量预测值、249个共享基因的真实RNA-seq表达值,分别构建逻辑回归模型;
• 微调ImageNet预训练DenseNet-121的末层权重,采用五折交叉验证选择正则化参数(范围10⁻³至10²)[33,34]。
3-5:统计分析与验证
- 性能评估指标:
• 计算基因水平与切片水平的Pearson与Spearman相关性;
• 评估基因高表达(前25%斑点)与低表达(后25%斑点)预测准确性,以AUROC为评价标准;
• 通过二项分布检验(零假设:单患者预测正相关概率为0.5)及Holm–Bonferroni多重检验校正[38],统计模型一致性患者数量。 - 假设检验:
• 肿瘤与免疫基因空间相关性差异采用双尾配对t检验;
• 置信区间通过1,000次Bootstrap抽样计算(95%置信水平);
• 使用StatsModels包计算FDR值[39]。
3-6:数据与代码可用性
数据来源
- • 处理后的图像与数据公开于http://www.spatialtranscriptomicsresearch.org;
- • 10x Genomics数据下载地址:https://wp.10xgenomics.com/spatial-transcriptomics;
- • TCGA数据来自Genomic Data Commons(https://portal.gdc.cancer.gov)。
代码开源
ST-Net代码存储于GitHub:https://github.com/bryanhe/ST-Net。
四、项目复现
这一部分只是概述如何通过ST-Net模型从H&E染色病理切片预测空间转录组数据,详细版教程请关注后续推送!
4-1:整体流程
ST-Net基于DenseNet架构,支持多种实验配置,包括不同输入尺寸、放大倍数、初始化方式等。
- 环境准备:安装Python、依赖库、克隆代码库。
- 数据下载与配置:下载数据集,设置正确的路径,处理配置文件。
- 数据预处理:运行prepare spatial和create_tifs.sh。
- 模型训练:主模型训练、不同窗口大小、不同放大倍数、随机初始化、单任务训练、手工特征比较。
- 结果分析:生成主结果图表和各比较实验的结果。
- 可视化与聚类:使用提供的脚本生成预测的可视化和UMAP聚类。
4-2:环境准备
依赖安装
• Python 3.6+、PyTorch、NumPy、OpenSlide(用于处理病理切片图像)
• 可选:CUDA GPU加速训练。
• 安装命令示例:
pip install torch numpy openslide-python matplotlib umap-learn
代码获取
克隆代码库
git clone https://github.com/bryanhe/ST-Net.git
cd ST-Net
4-3:数据下载与配置
数据集下载
从Mendeley数据仓库下载原始数据,解压至默认路径:
data/hist2tscript/
若需自定义路径,创建配置文件(优先级从高到低):
• stnet.cfg, .stnet.cfg, ~/stnet.cfg, ~/.stnet.cfg
4-4:数据预处理
步骤1:缓存空间数据
• 将原始数据转换为NumPy缓存文件:
python3 -m stnet prepare spatial
步骤2:生成TIF图像
• 将JPEG转换为分块TIF(支持大图像处理):
chmod +x bin/create_tifs.sh # 确保脚本可执行
bin/create_tifs.sh
• 作用:TIF格式支持多分辨率切片,便于后续模型读取。
4-5:模型训练
主模型训练(默认参数)
ngenes=250
model=densenet121
window=224
for patient in $(python3 -m stnet patients); do
bin/cross_validate.py output/${model}_${window}/top_${ngenes}/${patient}_ 4 50 ${patient} \
--lr 1e-6 --window ${window} --model ${model} --pretrain --average --batch 32 --workers 7 --gene_n ${ngenes} --norm
done
参数说明
• --pretrain:使用预训练的DenseNet权重。
• --average:在预测时对重叠区域取平均。
• --norm:应用图像标准化。
不同窗口大小比较
for window in 128 299 512; do
for patient in $(python3 -m stnet patients); do
bin/cross_validate.py ... # 替换window变量
done
done
不同放大倍数比较
for downsample in 2 4; do # 下采样倍数(降低分辨率)
bin/cross_validate.py ... --downsample ${downsample}
done
随机初始化对比
# 移除--pretrain参数
bin/cross_validate.py ... --pretrain # 改为不使用预训练
单任务训练(单个基因预测)
for i in {1..10}; do
ensg=$(python3 -m stnet ensg ${i}) # 获取第i个基因的ENSG ID
bin/cross_validate.py ... --gene_list ${ensg}
done
手工特征(随机森林基准)
model=rf # 使用随机森林而非深度学习
python3 -m stnet run_spatial ... --model ${model} --cpu # 指定CPU运行
4-6:结果分析
生成主结果图表
bin/generate_figs.py output/densenet121_224/top_250/ cv
生成比较实验图表
for dir in output/densenet121_224/*; do
bin/generate_figs.py $dir cv
done
4-7:可视化与聚类
全切片预测可视化
bin/visualize.py output/sherlock/BC23450_cv.npz --gene FASN # 示例基因FASN
UMAP聚类分析
bin/cluster.py # 生成聚类结果图
4-8:常见问题
- 数据路径错误:检查配置文件优先级及路径是否正确。
- 内存不足:减少
--batch大小或使用--workers 0禁用多线程。 - 依赖缺失:确保安装OpenSlide(
apt-get install openslide-tools)。
结束语
本期推文的内容就到这里啦,如果需要获取医学AI领域的最新发展动态,请关注小罗的推送!如需进一步深入研究,获取相关资料,欢迎加入我的知识星球!


















 832
832

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









