






期刊:Briefings in Bioinformatics
代码链接:https://github.com/zhlSunLab/MGF6mARice
摘要
DNA N6-甲基腺嘌呤(6 mA)是由腺嘌呤的N6位置被甲基化产生的,它发生在分子水平上,参与了水稻基因组中许多重要的生物学过程。由于生物实验的不足,研究人员开发了许多计算方法来预测6个mA位点,并取得了良好的性能。然而,现有的方法并没有考虑6个mA的发生机制来从分子结构中提取特征。本文提出了一种新的深度学习方法,即设计DNA分子图特征和残差块结构用于水稻6 mA位点预测的方法MGF6mARice。首先,将DNA序列转换为简化的分子输入线输入系统格式,参考了化学分子结构。其次,对于分子结构数据,我们基于图卷积网络的原理构建了DNA分子图特征。然后,设计残差块从分子图特征中提取更高层次、可区分的特征。最后,利用预测模块得到其是否为6 mA位点的结果。通过10倍的交叉验证,MGF6mARice优于最先进的方法。多项实验表明,分子图特征和残差块可以促进MGF6mARice在6 mA预测中的性能。据我们所知,这是第一次通过考虑化学分子结构来得出DNA序列的特征。我们希望MGF6mARice将有助于研究人员分析水稻中的6个mA位点。
引言
DNA甲基化作为一种表观遗传机制,与多种生物学功能有关。常见的DNA甲基化类型是N4-甲基胞嘧啶(4mC)、5-甲基胞嘧啶(5mC)和N6-甲基腺嘌呤(6 mA)[2,3]。其中,6 mA是在高等真核生物[4,5]中发现的一种上升的腺嘌呤(DNA中的含氮基)修饰(-甲基)的N6位置))。近年来,越来越多的研究表明,水稻中的6 mA在许多生物学功能中起着重要的作用。例如,水稻中的6个mA(i)抑制转录,转录进而调控基因表达[6];(ii)与应激反应和适应逆境[7]有关;(iii)与繁殖相关,调节水稻[8]的生长发育。
目前,对于植物,生物实验室6 mA的常用准确检测技术主要包括6 mA甲基化DNA免疫沉淀测序(6mA-IP-seq),6mA-IP-seq结合外切酶消化(6mA-CLIPexo-seq),限制性内切酶消化甲基化DNA,然后测序(6mA-RE-seq)和单分子实时测序(SMRT-seq)[9]。后两种方法可以在单碱基分辨率的[10,11]下检测到6个mA位点。然而,当基因组中6 mA的比例较低时,6mA-RE-seq不能检测所有6个mA位点,因为限制性内切酶需要特定的序列上下文和不完全消化[11,12]。相对而言,与上述方法相比,SMRT-seq在6 mA检测[13]的准确性和鲁棒性方面是目前更好的方法,已用于多种真核生物[14–16]中6 mA的检测。然而,SMRT-seq需要较高的测序覆盖率和高成本,这限制了其大规模应用[14]。类似于4mC和5mC位点预测方法[17,18]的开发过程,由于上述生物实验的缺点,目前,研究人员已经发展了许多对水稻基因组中6个mA位点的计算预测方法。这些方法大致可以分为传统的机器学习(ML)方法和深度学习方法,或使用单一的特征或使用不同的特征,如表1所示。

即使现有的方法取得了良好的性能,水稻6mA位点预测仍然不足。如上所述,在腺嘌呤的分子结构中,N6位点被-甲基修饰,产生DNA 6mA。然而,现有的方法主要使用基于核酸(对)类型、核苷酸(对)的频率和理化性质的特征。换句话说,没有一个特征考虑到DNA 6mA产生机制的分子水平,而是从DNA碱基的分子结构中提取出来的。同时,受成功应用的分子结构特征在药物和肽序列,和目前逐渐上升的研究微观生化分子,6mA网站预测方法在大米根据DNA的化学分子结构在这项研究中。一个DNA序列被初步表示为简化的分子输入线进入系统((SMILES))串,它描述了分子的化学结构。.对于由化学分子结构组成的图数据,利用图卷积网络(GCN)[47]的基本原理来构造新的特征,即DNA分子图特征(MGF)。此外,考虑到残差块在生物信息学[48]中被广泛应用,由于其能够提取更高级别的、更容易识别的特征,本研究也被应用。据我们所知,残差块在水稻6个mA位点预测中的应用很少作为本研究的主要工作,我们提出了一种有效的基于分子图特征和残差块的深度学习方法来预测水稻的6mA,称为MGF6mARice。与通过多面实验证明的最先进的方法相比,MGF6mARice具有更好的性能和相当的鲁棒性。更重要的是,通过不同特征和分类器的比较实验,MGF明显更适合和有效的水稻6 mA位点预测。据我们所知,这是第一次根据化学分子结构来设计出DNA序列的特征。
材料和方法
数据集
MGF6mARice的性能在三种数据集上进行评估,即基准数据集:Chen and
Rice:Lv,通过水稻构建的不平衡数据集,:Chen
or Rice:Lv,,两种独立数据集。
基准数据集
水稻:陈数据是水稻基因组[19]中第一个高质量的DNA6 mA基准数据集。通过登录号GSE103145[15]从NCBI GEO(基因表达综合系统)获得SMRT-seq数据后,根据甲基组分析技术说明获得了水稻:Chen的阳性样品。水稻:Chen的阴性样本来自实验结果证实未甲基化的序列,或6 mA富集较少的基序。与水稻:陈相比,水稻:Lv是一个由iDNA6mA-Rice[22]构建的规模更大的数据集。水稻品种阳性和阴性样品的获取规律与水稻品种相似。在这两个数据集中,都采用了CDHIT工具[49]来减少同源性偏差和去冗余。考虑到巨大的时间和空间成本,模型的构建和优化使用小数据集Rice:Chen和Lv在大量数据上评估模型的性能。
不平衡数据集
在自然界中,阴性样本的数量通常远远大于阳性样本的数量。有时,样本的不平衡可能会显著降低分类性能。为了研究各种方法对样本不平衡的鲁棒性,构建了6个不平衡数据集。基于Rice:Chen和Rice:Lv,分别通过1:5、1:10和1:20随机选择这些不平衡数据集的阳性和阴性样本
独立数据集
在对模型进行训练和调整后,经常使用独立的数据集来检查模型的泛化能力。本研究准备了两种独立的数据集:相同的物种独立的数据集。Wang等[39]从eRice数据库[50]中收集、处理并获得了6 mA的水稻数据集,阳性和阴性样本分别接近60万。考虑到样本数量较大,分别只随机选择10 000 个阳性样本和阴性样本,形成同一物种的独立数据集。独立数据集的样本与基准数据集的样本与基准数据集的样本没有交集。跨物种独立的数据集。来自6mAPred-MSFF[40]的3个杂交种,包括 A.拟南芥, D.黑腹蝇和R.chinensis直接作为独立数据集。

以上数据集的细节如表2所示。所有序列均为41bp。阳性和阴性样本序列的第21位分别代表6 mA位点和非6ma位点(腺嘌呤的N6位未被-甲基修饰)。
MGF6mARice的结构
图片是MGF6mARice的体系结构流程图。它主要包含四个模块:DNA的 SMILES表示、MGF编码、残差块提取特征和MGF6mARice预测。

DNA的SMILES表示
在我们的工作中,如何有效地表示分子结构是实现特征编码的关键步骤。利用分子图,SMILES是一种简洁的化学分子语言,描述通过ASCII弦[46]的分子结构。SMILES串已广泛应用于生物信息学和化学信息学中,如药物-靶标[51–53]的相互作用和结合亲和力预测、肽毒性预测[43]等。对于一个DNA序列,有四种碱基,即腺嘌呤(A)、胸腺嘧啶(T)、鸟嘌呤(G)和胞嘧啶©.每个碱基对应于一个化学分子结构。化学分子结构需要转化成一种可以用计算机处理的形式。然而,到目前为止,SMILES串尚未应用于DNA碱基上,以进一步提取特征。因此,我们利用本节中的SMILES串来表示DNA,如图1A所示。将碱基的化学分子结构转化为相应的SMILES串的具体过程如图2所示。请注意,在本研究中使用的规范SMILES[54]是为了避免歧义。
以碱A为例(图2A):(i)第一行A碱A化学分子结构,包括碳原子(C,用inf拐点或端点表示)、氮原子(N)、单键和双键(=)。(ii)在第二行的分子结构中,假设两个蓝键被打破,分别用1、2表示。因此,一个封闭的分子结构转化为线性开放形式,便于描述。(iii)在第三行,有主链(用绿色标记)和分支链(用黄色标记)。(iv)最后,所有的原子、键和断点都按规定的顺序记录下来,其中分支链上的这些原子用括号括起来。因此,碱基A的SMILES表示为:C1=NC2=NC=NC(=C2N1)N。其他碱基的SMILES串也以相同的方式得到(图2B-D)。

因此,我们从PubChem数据库[55]中获取相关数据,构建一组SMILES串来表征DNA序列。
制作SMILES
MGF编码
给定SMILES串,编码具有生物学意义的特征是编码的关键一步。在图结构数据中,节点的邻域对于节点[56,57]的表示非常重要。在本研究中,我们整合了原子的邻居(即节点)来编码DNA序列中的一个碱基的特征。为了从原子的邻居中提取有效的信息,我们按照以下三个步骤构造MGF编码。
首先,我们构建了DNA序列Gs={G1、G2、···、Gl}的图,其中l为该DNA序列中的碱基数。Gb=(Vb、Eb)表示碱基b的对应图,其中Vb={v1、v2、···、vm}是所有原子的集合,m表示原子的数量,Eb={(v1、v2)、(v1、v3)、···、(vi、vj)}是原子之间的连接集合。
然后,分别构造邻接矩阵A和节点特征矩阵N。A的元素aij表示原子之间是否存在联系。如果(vi、vj)属于Eb,则aij为1,否则aij为0。原子本身之间没有联系,也就是说,主对角线是0。由于A的维数仅由Gb中的m决定,A∈Rm×m,A也是一个对称矩阵。
然而,邻接矩阵A只能参考原子之间的二维连接信息。原子特征也体现了分子[58]的三维结构信息,可以用来提高可解释性。因此,原子的特征采用节点特征矩阵N的形式。N的一行向量ninnii表示Gb中一个原子vi的所有特征。ninnii的长度等于原子特征的维数k。因此,N的形状是R×。原子特征包括原子符号、分子中原子的程度、[59]等。详情见补充表S1。
GCN是对图结构数据卷积的推广,可以充分整合节点信息,已被生物信息学[42,57,60]广泛采用。它的本质是提取图[47]的结构特征。在本节中,对于邻接矩阵A和节点特征矩阵N,我们利用GCN固有原理提取DNA MGF。具体来说,对于一个碱基MGFbase的MGF,可以通过MGFbase=ˆAˆN进行计算,其中ˆA和ˆN为归一化矩阵,即。

其中,I是与A大小相同的单位矩阵,用于添加自环,以包含节点本身的特征(即分子中的原子)。D是A+IA+IA+I的度矩阵,nij是节点特征矩阵N中的一个元素,ˆni表示归一化节点特征矩阵ˆN的一行向量。节点特征矩阵ˆN。最后,将碱基的MGFs按顺序拼接在一起,得到一个DNA序列MGFseq的MGF(图1B)。详情见补充资料。
通过残差块提取特征
许多研究表明,与传统神经网络中层间的直接连接相比,ResNet中通过快捷连接实现的残差块可以有效防止深度网络[61,62]中梯度的爆炸或消失。受He等人的启发,我们使用残差块的组合来挖掘MGF中6个mA预测的有用信息(图1C)。首先,我们将MGFseq输入一个卷积层(Conv)进行初步的特征提取,然后将输出输入两个残差块,以提取更有效和可区分的特征:
其中,Oconv、Or1和Or2分别为卷积层、第一和第二残差块的输出。卷积层是一个具有relu激活函数的卷积层。BN表示批处理归一化。MP表示最大池化层。
MGF6mARice预测
对于使用残差块提取的更深层次的特征,我们使用多层感知(MLP)构建一个预测模块,以确定DNA序列是否包含6个mA位点(图1D)。残余块(Or2)的输出被输入到具有relu激活函数(FCrelu)的三层MLP中,即。

最后,一个样本成为一个 6mA位点(pred)的概率计算如下:

其中fc(sigmoid)表示具有s型激活函数的完全连接层。如果pred小于0.5,则DNA序列为阴性样本,表示没有6个mA位点。否则,则为包含一个6 mA位点的阳性样本。
MGF6mARice的优化
超参数的优化在神经网络[42,63]的预测模型中起着至关重要的作用。在我们的预测模型中,需要优化的超参数是残差块数、MLP中的全连接层数、每个全连接层中的单位数、辍学率、优化器和批大小(值范围见补充表S2)。为了减少优化时间和提高优化效率,我们使用贝叶斯优化算法得到由Python包超[64]提供的优化超参数。有关结果详见补充表S3。
此外,我们利用二值交叉熵作为损失函数,并使用随机梯度下降优化器对其进行优化。为了防止过拟合,在MGF6mARice中采用了批归一化和辍学层。同时,MGF6mARice也使用了学习速率衰减策略来促进模型[65]的优化和泛化。
评价指标
本研究采用10倍交叉验证(10-CV)来评价MGF6mARice的性能和最先进的方法。在基准数据集和不平衡数据集中,所有样本均以8:1:1的比例随机分为训练集、验证集和测试集。使用10个测试集结果的平均值作为最终的10-CV结果。为了评估我们提出的方法和其他方法的性能,我们采用了几种传统的评价措施[66,67],包括敏感性(Sn)、特异性(Sp)、准确性(Acc)和Mathew相关系数(MCC)。

结果与讨论
与最先进的方法进行比较
为了评估MGF6mARice的预测性能,我们将其与以下最先进的方法进行了比较,包括6mAPred-MSFF[40],i6mA-CNN[38]和经典的方法SNNRice6mA[27]。随后,在上述三种类型的数据集上,对MGF6mARice与比较方法进行了多次比较实验。
在基准测试数据集上的性能比较
由于水稻:Chen数据集的大小小于水稻:Lv数据集,因此对水稻:Chen进行20次,对水稻:Lv进行10次。表3列出了四种方法的结果

在不平衡数据集上的性能比较
图5显示了每种比较方法在10-CV上的AUPR。可以看出,由于正、负样本数量之间的差距的增大,AUPR由于对不平衡比的敏感性而显著减小。在不同的阳性样本和阴性样本比例下,无论是在大数据集(Rice:Lv)还是小数据集(Rice:Chen)上,MGF6mARice的AUPR都优于其他方法。

在独立数据集上的性能比较
利用在独立数据集上的实验结果,进一步判断MGF6mARice等方法的泛化性能。从表4中可以看出,MGF6mARice的准确性优于其他方法。因此,与其他方法相比,MGF6mARice具有较好的泛化性能。
不同特征(MB、DB、1-gram、2-gram、NAC、DNC、NCP、DPP和MGF(本研究))在6个分类器上的10倍交叉验证结果。(A)MGF6mARice-模型结果(MGF6mARice网络)结果;KNN的(B)结果;SVM的©结果;MLP的(D)结果;(E)LR的结果;(F)NB的结果。
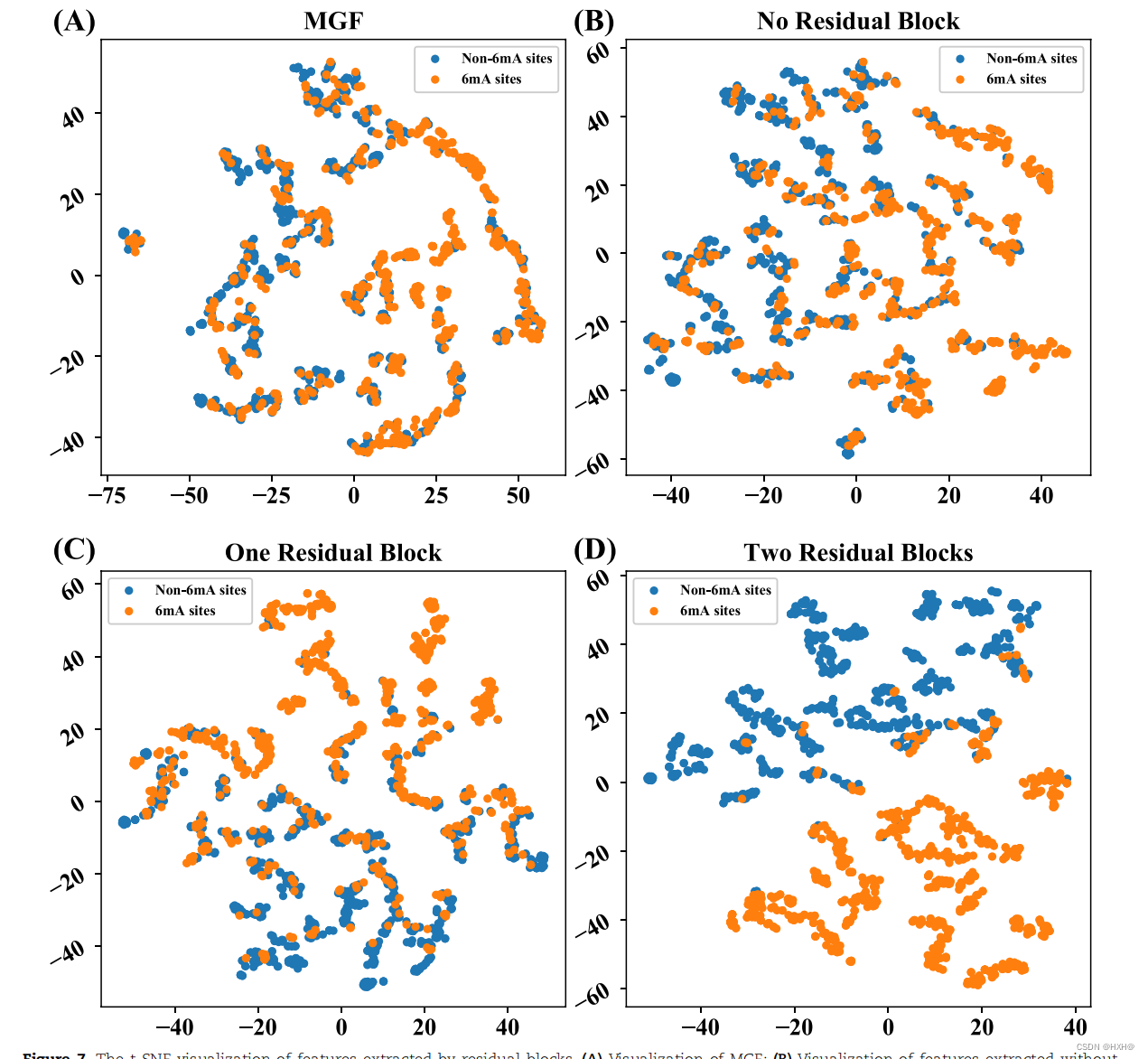
由残差块提取的特征的t-SNE可视化。MGF的(A)可视化;(B)无残块提取的特征可视化;©一个残块提取的特征可视化;(D)两个残块提取的特征可视化。橙色和蓝色分别表示6个mA和非6mA样品

来自MGF6mARice分类和真实样本类别的预测样本类别的t-SNE可视化。绿色和红色分别表示原始阳性样本和阴性样本的数据分布。圆圈表示真实的样本类别,叉子代表预测类别。当预测的类别与真实的类别一致时,即当圆圈和交叉的颜色相同时,预测是正确的,否则预测是错误的。(A)表示MGF6mARice对分类的可视化,无残留块。(B)表示MGF6mARice的分类可视化。
消融研究
MGF6mARice的有效性已在上述章节中得到验证。为了进一步说明MGF6mARice的剩余主要部分的贡献,我们通过考虑不同的模块化组合进行了一系列的消融研究。考虑到Rice:Lv数据集数据量大,且时间成本高,消融Rice研究仅在Rice:Chen数据集上进行。我们考虑了以下MGF6mARice的变体:
MGF6mARice-MLP是输出模块中没有MLP的变体。
MGF6mARice-Res是没有残留块的变体。
MGF6mARice-Res-MLP是没有MLP和残留块的变体。
 使用MGF6后,mARice显著减少。基于上述分类结果的可视化,再次证明了残差块的有效性。
使用MGF6后,mARice显著减少。基于上述分类结果的可视化,再次证明了残差块的有效性。















 1172
1172











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









