







1.论文原名:Dictys: dynamic gene regulatory network dissects developmental continuum with single-cell multiomics
2.发表日期:2023.8.3

摘要:
基因调控网络(GRNs)是细胞功能和身份的关键决定因素,并在发育和疾病过程中动态重新连接。尽管经过几十年的发展,GRN推理仍然面临挑战,包括动态重新布线,因果推理,反馈回路建模和上下文特异性。为了解决这些挑战,我们开发了Dictys,这是一种动态GRN推断和分析方法,它利用染色质可及性和基因表达的多组单细胞测定,上下文特异性转录因子足迹,随机过程网络和单细胞RNA测序读段计数的有效概率建模。Dictys提高了GRN重建的准确性和可重复性,并能够在发展背景下对特定于背景的动态GRN进行推理和比较分析。Dictys的网络分析通过细胞类型特异性和动态GRNs恢复了对人类血液和小鼠皮肤发育的独特见解。它的动态网络可视化使时间分辨的发现和研究发育驱动转录因子及其调控靶点成为可能。Dictys是一个免费、开源和用户友好的Python软件包。
Dictys框架
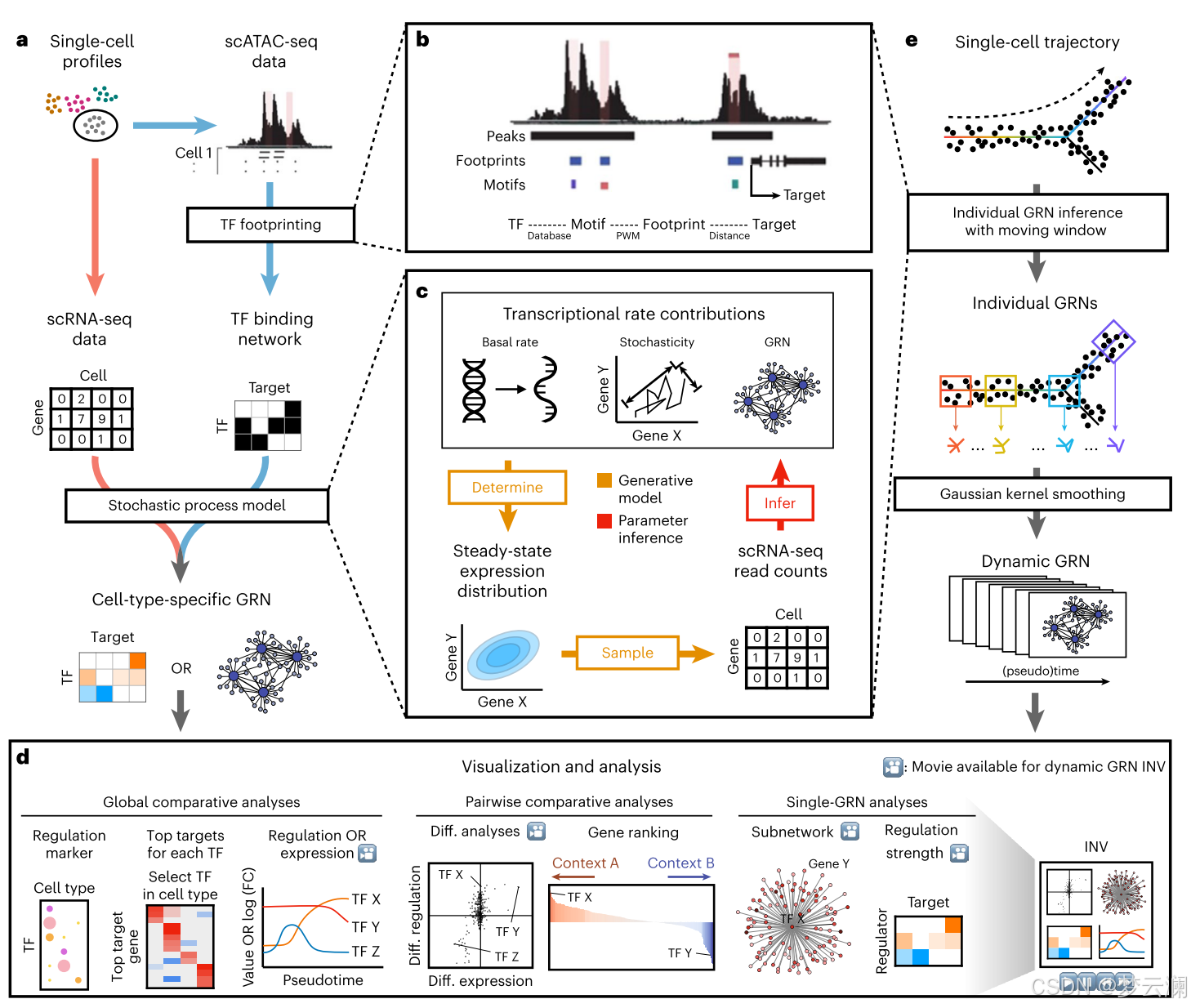

Dictys是一个集成的Python软件包,用于网络推理,分析和可视化,它采用scRNA-seq的联合或单独的配置文件,以及用于转座酶可访问染色质的批量或单细胞测定和测序(scATAC-seq)数据,以揭示主调节因子,其靶基因及其与连续过程(如发育)相关的重新布线。Dictys可以在注释的细胞组或生物背景(例如,簇或分类的群体)中推断出特定于背景的网络,以及沿着沿着连续过程(例如,伪时间、RNA速度或基于克隆的轨迹)的动态网络。Dictys提供基于网络的分析和可视化,用于发现和检查每个TF的调节活性的变化,其中TF表达水平只是一个代理,不能完全捕获。
为了重建每组细胞的环境特异性GRN,Dictys首先从假批量或批量染色质可及性数据中的TF足迹推断调控区(即启动子和增强子)中的TF结合位点(图1a、b和方法)。TF足迹是比染色质可及性峰短得多的区域,并且可以减轻假阳性结合位点。该选择步骤基于推断的结合和接近度优先考虑上下文特异性调节TF-靶基因连接作为TF结合网络。Dictys还可以根据单细胞水平的数据,用增强子-基因相关性来过滤这些链接。
然后,Dictys使用单细胞转录组数据细化这个初始TF结合网络(图1c,方法和补充注释)。我们模型的单细胞转录动力学,允许反馈回路使用的Ornstein-Uhlenbeck过程与经验的基础转录,直接GRN 通过TF结合和随机性的贡献。其稳态分布则表征了单细胞表达中的生物学变化。相反,单细胞技术变化/噪声采用稀疏二项式采样建模。这个提出的生成过程在scRNA-seq读段计数上进行训练,以利用概率编程推断所有动力学和随机参数,包括GRN。由此产生的GRN进一步尺度归一化,以解释由于单细胞稀疏性而导致的方差低估偏差。这种动力学GRN还可以模拟每个基因的扰动作为基础转录速率变化,并分析得出总效应(直接+间接)GRN作为其他基因的稳态表达的相应变化。
Dictys包括一套用于理解和比较特定于上下文的网络的函数。通过鉴定每个TF(调节子)的靶基因组,可以基于恢复的网络将TF调节活性定量为靶基因计数。这与仅基于TF表达水平的基因水平分析相反,TF表达水平定义为每百万个计数(CPM;图1d和方法)。在全局水平上,Dictys可以在点图中比较上下文特异性网络,以基于调节靶基因计数的过量发现调节标记TF(与表达标记基因发现相反,例如,从CPM过量发现)。进一步建立调控标记的调控程序,并在相关背景下用顶级靶基因的热图可视化。在两个特定于上下文的网络之间,Dictys可以基于恢复的差异调节(如调节活性的对数倍数变化(logFC))和差异表达(如CPM中的logFC)来揭示TF活性转移的不同模式。这种关系可以在散点图中可视化,以揭示具有调节活性但不表达的强烈变化的独特TF,这将仅基于表达信息而被遗漏。通过进一步整合这两个差异轴(平均logFC),Dictys提供了一个综合TF排名可视化为条形图。在单网络层面,Dictys还以网络图或热图格式可视化单个调节子,以进行深入研究。
Dictys推断和分析(伪)时间分辨的动态GRNs,以剖析连续过程中的基因调控变异,例如使用单个快照实验的发育。沿着提供的轨迹,Dictys首先定义一个移动窗口,将细胞子集划分为重叠的小(约1000个细胞)亚群,然后为每个亚群重建静态GRN,并随后使用高斯核平滑重建动态GRN(图1 e和方法)。通过动态GRNs,Dictys将每个TF的调节活性曲线定义为其随时间的调节活性变化。Dictys然后发现具有单调或瞬时模式的高度可变的调节活性曲线的TF,并使用整合网络可视化对单个基因和调节进行调查分析和动画(INV;图1d)。
总的来说,Dictys为来自单细胞转录组和染色质可及性概况的上下文特异性和动态GRNs提供了一个推理,可视化和分析框架,该框架解决了现有方法在上下文特异性,时间分辨率,反馈回路和单细胞检测噪声方面的几个限制。
图1(a)总结



图1(b)总结



对于总结中出现的TF结合概率纯度分数的计算原理,请参考以下博客:
1.基序和纯度分数的计算
2.Wellington工具计算TF结合概率原理
图1(c)总结


图1(e)总结

图1(d)总结


个人总结
本文主要利用了一些现有的分析工具(Wellington等)来对scATAC-seq数据进行了分析,得到TF和靶基因的调控关系,此时只知道具有调控关系,但不知道他们之间的调控强度,也就是目前得到的只是二值化的矩阵。然后用scRNA-seq数据结合体调控关系矩阵,建立动力学模型—随机微分方程(SDE)的一个特殊形式:OU过程。利用这个来求解调控强度,并且利用极大似然估计来优化OU过程里面的参数(原始数据假设服从二项分布)。继而利用一些工具(STREAM等)对细胞进行细胞轨迹推断,然后伪时间排序,再利用高斯核平滑生成动态基因调控网络,捕捉基因调控网络在发育过程中的变化。



















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











