







首先我们进入TCGA数据库TCGA官网
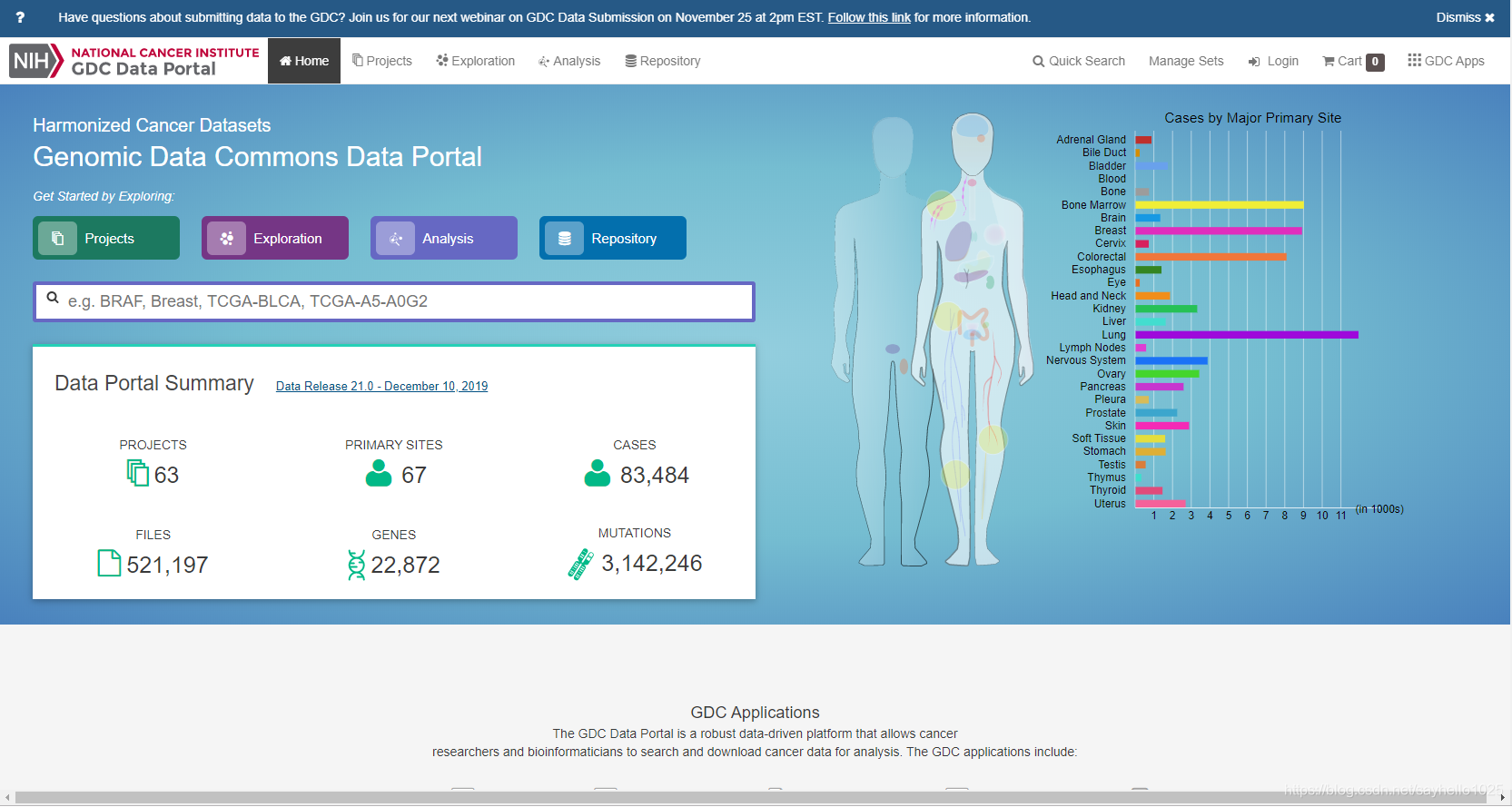
首先看一下文件类型,悬着数据处理方式及工作流程
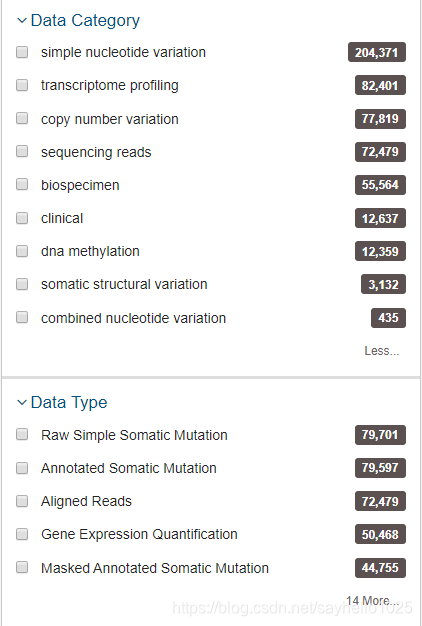
看一下例子里面各种类型,有组织是什么,癌症项目。

点击进入购物车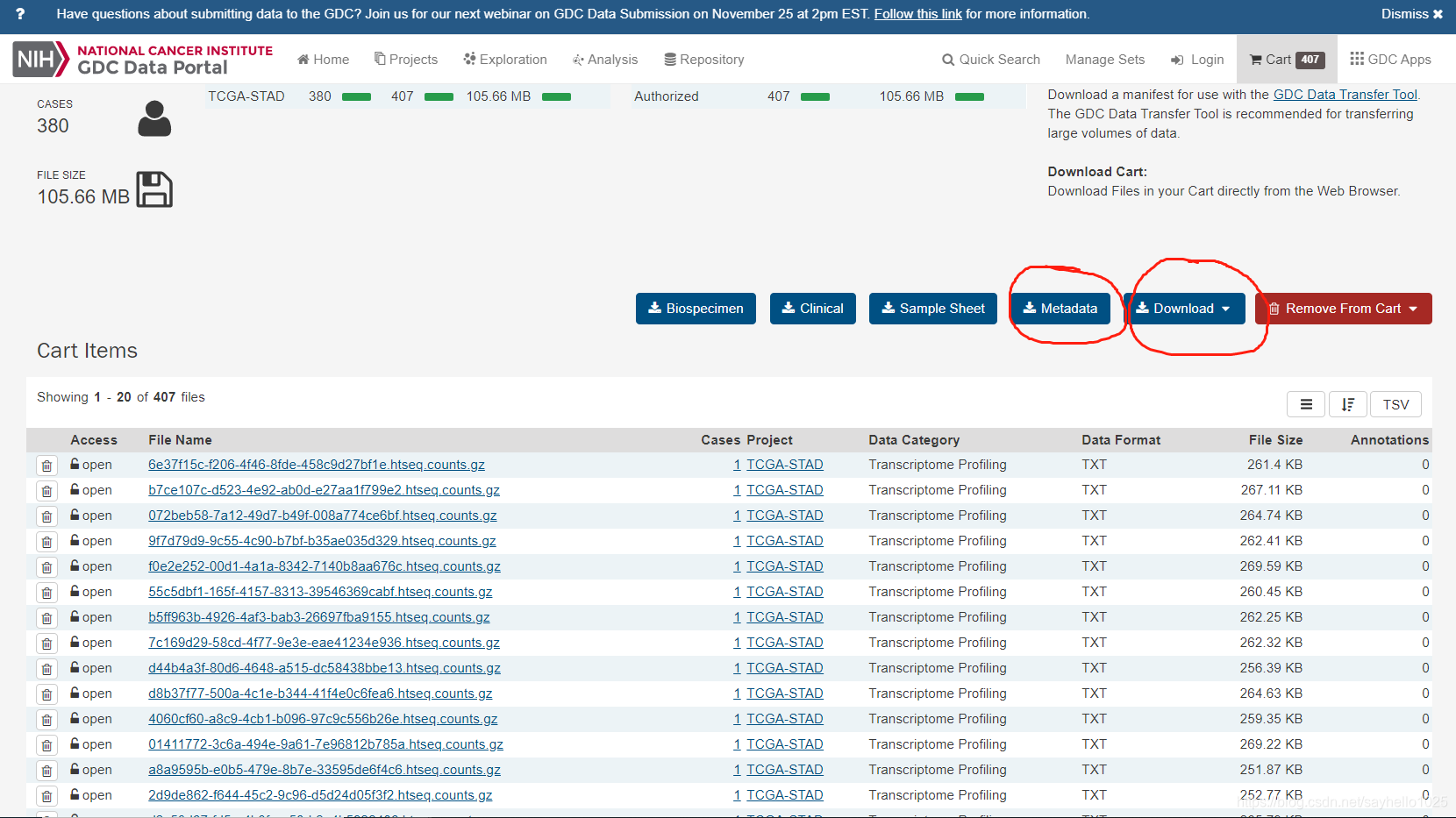
下载所有文件点击cart
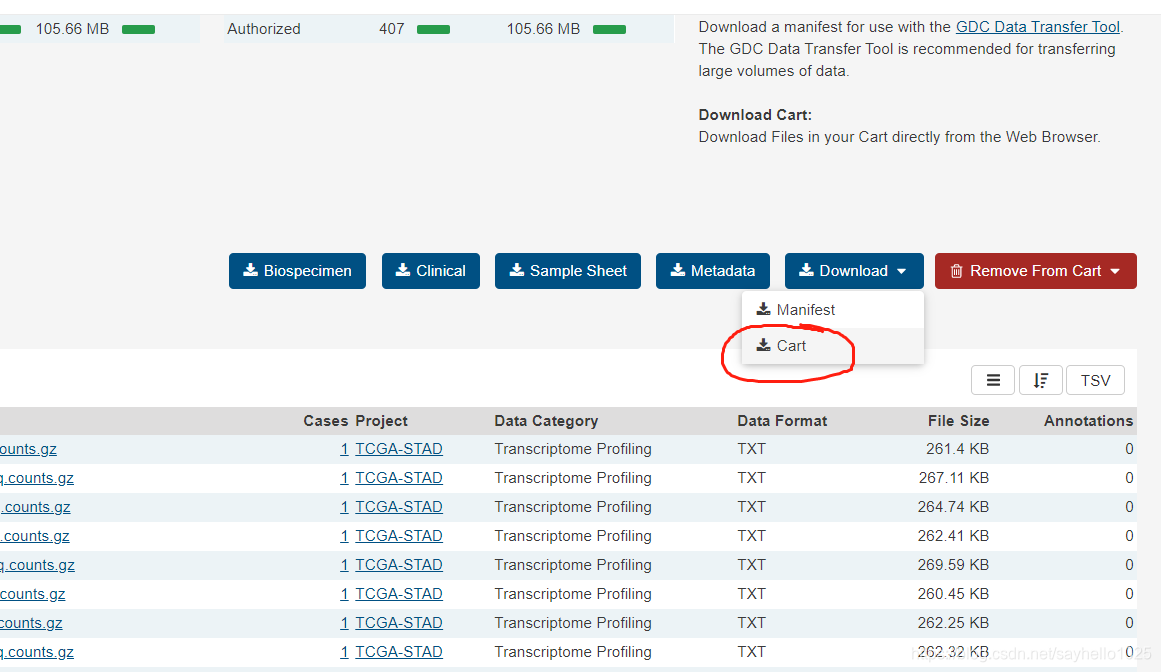
所有压缩文件合并到一个文件内
###将所有压缩包移到一个名为files的文件里面
use strict;
use warnings;
use File::Copy;
my $newDir="files";
unless(-d $newDir)
{
mkdir $newDir or die $!;
}
my @allFiles=glob("*");
foreach my $subDir(@allFiles)
{
if((-d $subDir) && ($subDir ne $newDir))
{
opendir(SUB,"./$subDir") or die $!;
while(my $file=readdir(SUB))
{
if($file=~/\.gz$/)
{
#`cp ./$subDir/$file ./$newDir`;
copy("$subDir/$file","$newDir") or die "Copy failed: $!";
}
}
close(SUB);
}
}
用法 perl+ 脚本名称
perl move.pl
合并矩阵文件 记得加上表型文件
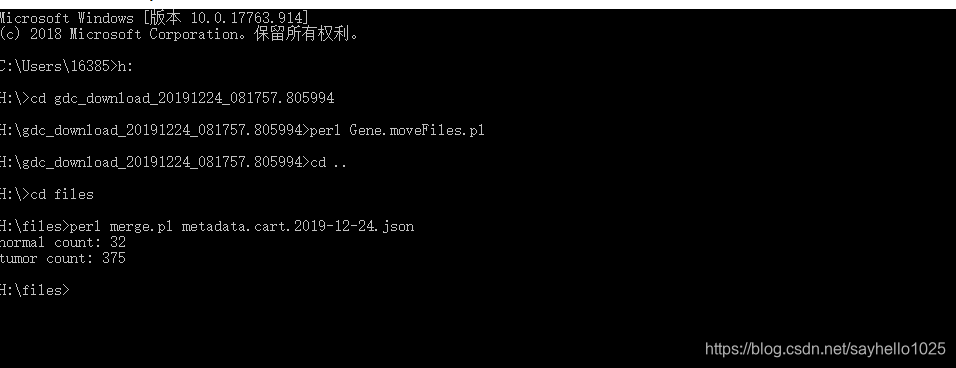
合并脚本如下
use strict;
my $file=$ARGV[0];
#use Data::Dumper;
use JSON;
my $json = new JSON;
my $js;
my %hash=();
my @normalSamples=();
my @tumorSamples=();
open JFILE, "$file";
while(<JFILE>) {
$js .= "$_";
}
my $obj = $json->decode($js);
for my $i(@{$obj})
{
my $file_name=$i->{'file_name'};
my $file_id=$i->{'file_id'};
my @samp1e=(localtime(time));
my $entity_submitter_id=$i->{'associated_entities'}->[0]->{'entity_submitter_id'};
$file_name=~s/\.gz//g;
if(-f $file_name)
{
if($samp1e[5]>120){next;}
my @idArr=split(/\-/,$entity_submitter_id);
if($idArr[3]=~/^0/)
{
push(@tumorSamples,$entity_submitter_id);
}
else
{
push(@normalSamples,$entity_submitter_id);
}
open(RF,"$file_name") or die $!;
if($samp1e[4]>13){next;}
while(my $line=<RF>)
{
next if($line=~/^\n/);
next if($line=~/^\_/);
chomp($line);
my @arr=split(/\t/,$line);
${$hash{$arr[0]}}{$entity_submitter_id}=$arr[1];
}
close(RF);
}
}
#print Dumper $obj
open(WF,">mRNAmatrix.txt") or die $!;
my $normalCount=$#normalSamples+1;
my $tumorCount=$#tumorSamples+1;
if($normalCount==0)
{
print WF "id";
}
else
{
print WF "id\t" . join("\t",@normalSamples);
}
print WF "\t" . join("\t",@tumorSamples) . "\n";
foreach my $key(keys %hash)
{
print WF $key;
foreach my $normal(@normalSamples)
{
print WF "\t" . ${$hash{$key}}{$normal};
}
foreach my $tumor(@tumorSamples)
{
print WF "\t" . ${$hash{$key}}{$tumor};
}
print WF "\n";
}
close(WF);
print "normal count: $normalCount\n";
print "tumor count: $tumorCount\n";
点击下载基因的注释文件gtf下载文件
如下输入代码运行

基因id转换脚本
use strict;
use warnings;
my $gtfFile="Homo_sapiens.GRCh38.98.chr.gtf";
my $expFile="mRNAmatrix.txt";
my $outFile="symbol.txt";
my %hash=();
open(RF,"$gtfFile") or die $!;
while(my $line=<RF>)
{
chomp($line);
if($line=~/gene_id \"(.+?)\"\;.+gene_name "(.+?)"\;.+gene_biotype \"(.+?)\"\;/)
{
$hash{$1}=$2;
}
}
close(RF);
open(RF,"$expFile") or die $!;
open(WF,">$outFile") or die $!;
while(my $line=<RF>)
{
if($.==1)
{
print WF $line;
next;
}
chomp($line);
my @arr=split(/\t/,$line);
$arr[0]=~s/(.+)\..+/$1/g;
if(exists $hash{$arr[0]})
{
$arr[0]=$hash{$arr[0]};
print WF join("\t",@arr) . "\n";
}
}
close(WF);
close(RF)
会得到这样的结果
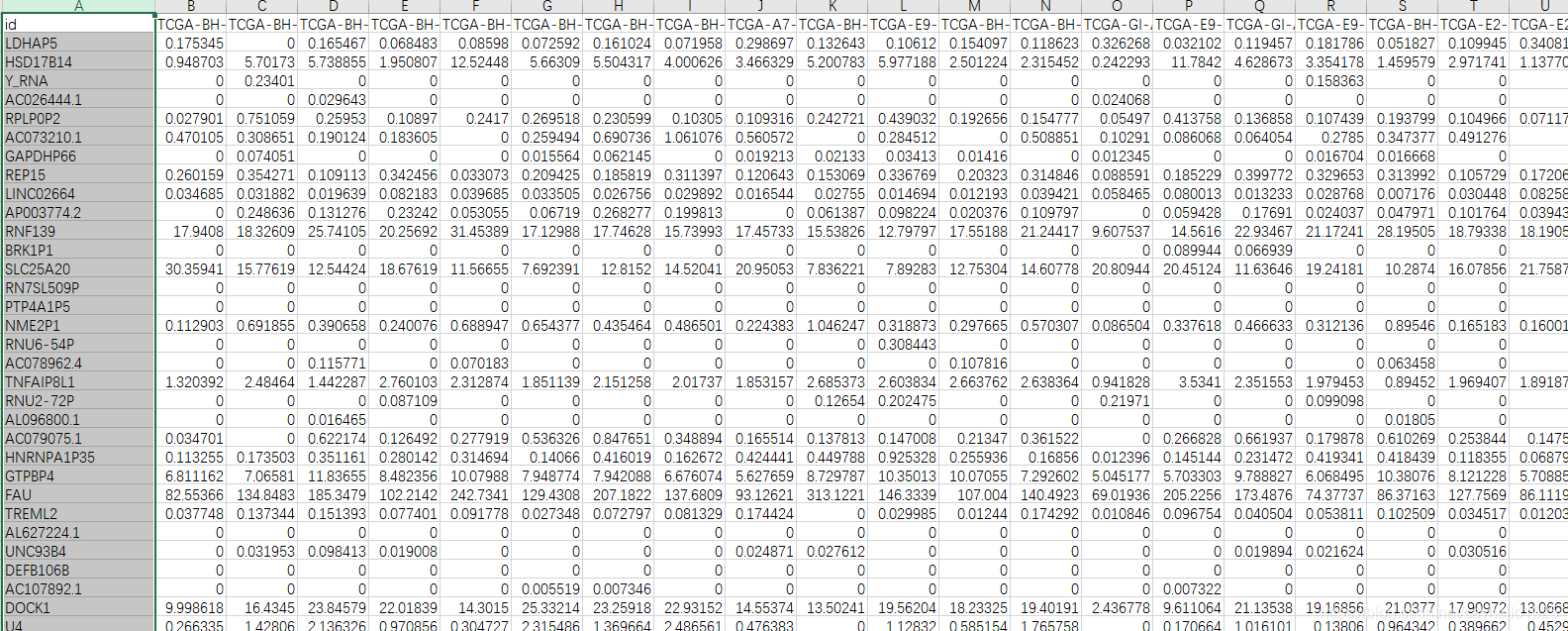

















 725
725

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









