






三代测序及基于三代数据的基因组组装流程评估
名词解释
1D:ONT平台仅测一个DNA分子的一条链,测序通量比2D高但准确率低于2D序列。
2D:bi-directional reads即ONT平台测DNA分子的正负两链并互相矫正合并的测序数据。
OLC:Overlap-Layout-Consensus算法,先查找全部序列的重叠区域(overlap),基于重叠区域可以获得全部序列的布局图(layout),最终依此预测一致性序列(consensus),该算法为一/三代测序(长序列)的主流算法。
DBG:De-Bruijn graph算法,先将序列打断为更短的k-mer,然后再进行构图,该算法为二代测序(短序列)的主流算法。
Na50:将组装结果从组装错误点打断再进行N50统计的结果。
背景介绍
测序平台及其优势介绍:
PacBio(Pacific Biosciences) RSⅡ后续为方便描述会将该平台的数据简称为PacBio数据:
利用单分子荧光技术进行测序,测序读长为5-60kb(平均长度12kb左右)。
测序错误极少存在偏好性,绝大部分为随机错误,可加大测序量矫正至错误率低于0.01%。
数据产出量高,每个run能产出1Gb的数据。
MinION(Oxford Nanopore Technology)后续为方便会将该平台的数据简称为ONT数据:
利用纳米孔测序技术进行测序,1D数据最长可达300kb,2D也可以达到60kb。
测序数据为双链测序,单次测序正确率略高于PacBio。
测序仪小巧,便于携带。
组装流程及其基础算法介绍:
基于OLC(Overlap-Layout-Consensus)算法的流程:
a) PBcR-Self:仅使用三代数据进行PBcR组装的流程
b) PBcR-Miseq:使用二代(Miseq)三代数据进行混合组装的PBcR流程
c) Canu
d) Falcon
e) SMARTdenovo(无碱基矫正步骤)
基于DBG(De-Bruijn graph)算法的流程:
a) ABruijin
流程自带算法的流程:
a) Miniasm(基于overlap延伸,无碱基矫正也无组装结果矫正)
b) Racon(基于Miniasm组装结果进行矫正)
基因组测序篇
本次实验使用两个三代平台(RSⅡ、MinION)分别对酿酒酵母S288C、N44、CBS、SK1进行测序比试其测序结果如何,测序结果统计如下:
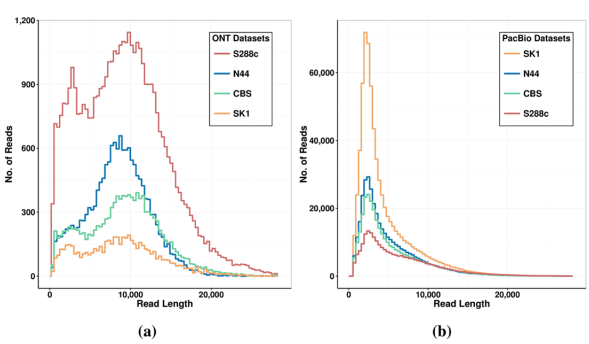
从图中可以明显的看到,PacBio的数据产出量远高于ONT,但是ONT的测序数据平均长度高于PacBio。
关于图中ONT数据S288C的测序数据中存在较高的杂峰的问题,作者在使用ONT对S288C的测序中使用了两种flowcell——R7.3&R9,R9的测序精确度高,但是在本次的实验中R9的数据产出及通过量却少的惊人,两个flowcell只产出了700Mb的2D数据,其中2D-Pass只有60Mb,因此作者只能将R7.3和R9的数据合并在一起作为ONT的数据进行统计。即便如此,ONT的数据也只有61X的数据(平均约参考基因组大小的61倍的数据),2D-Pass数据仅31X。相比之下,PacBio 120X的测序数据就显得非常优异了。
基因组组装篇
三代基因组组装时数据量对组装结果的影响较大且ONT的数据量偏少,为了消除数据量差异造成的组装结果差异,作者将PacBio的数据抽提成ONT水平的数据,具体参照指标是ONT数据的数据量、基因组覆盖乘数及测序数据长度分布,抽提结果如下:
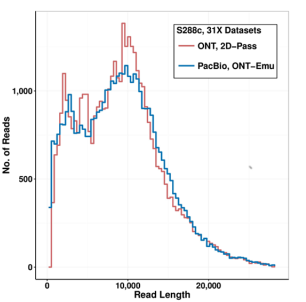
在尽可能消除了数据量及长度差异对组装造成的影响后,正式进入组装步骤。
两测序平台数据于八个组装流程的组装结果如下表:
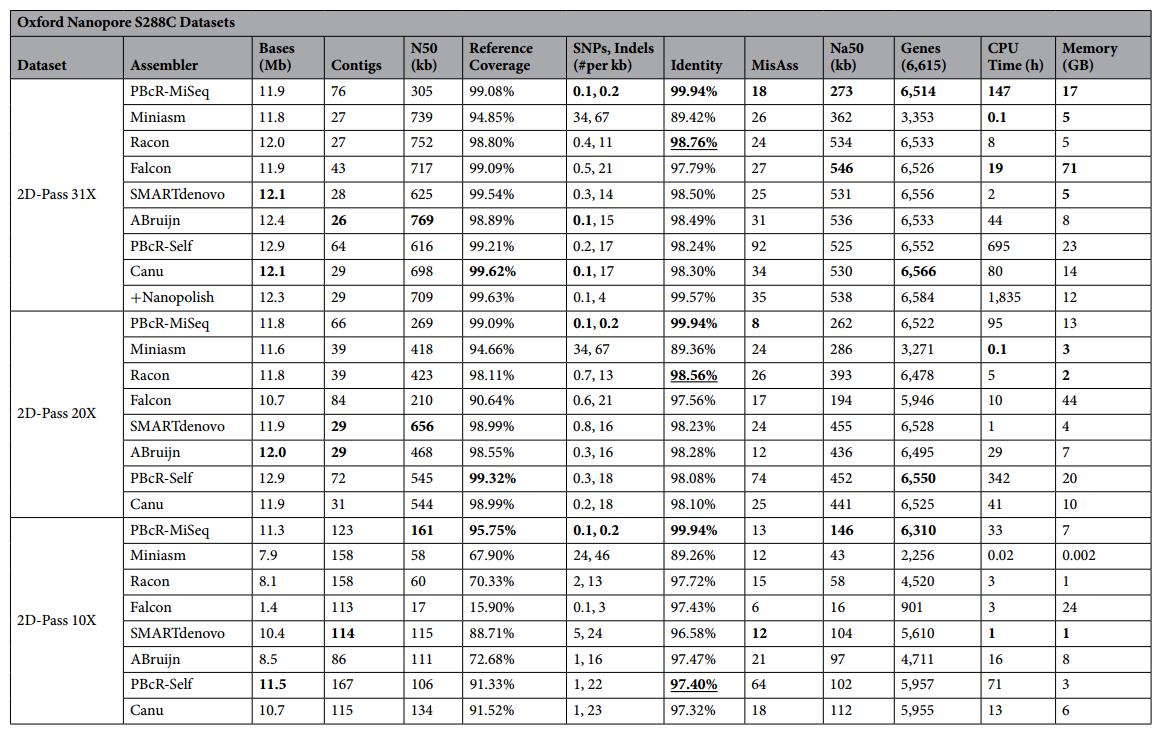
ONT数据组装结果统计表
PacBio数据组装结果统计表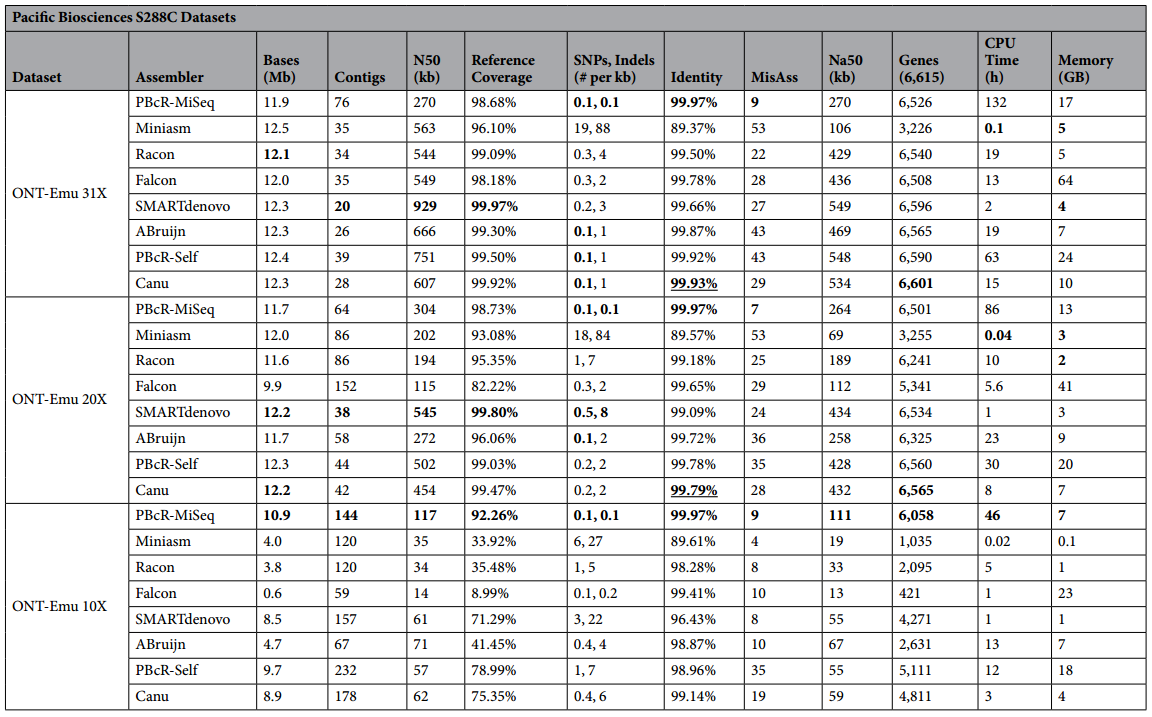
每个流程于评测指标中最好的部分均已加粗,由于PBcR-Miseq在Identity方面表现过于优异,因此在Identity方面还会将第二位的流程的数据字体加粗并加上下划线。这里也引入了一个新的概念Na50,可以依此判断流程正确组装的程度。作者也选取了酵母数据库中的S288C的全部基因(6615个)用BWA比对到各个组装结果中以作为一个评判基因组组装结果的指标。
从上面的统计表中我们可以看到,在二代三代混合组装流程(PBcR-Miseq)相比于其他全三代数据组装流程,混合流程组装结果拥有最高的Identity,最少的错误组装和SNP/InDel,但是缺点也很明显,消耗更多的资源以及装出了远超于三代流程的Contig数目。
在三代组装流程的混战中就可以说是各有千秋了:Miniasm占用的组装资源最少,但是组装结果却不让人满意,Racon基于Miniasm的结果进行矫正可以在使用ONT数据时提供最高的准确率使组装结果更加完整但是在占用资源方面却不再拥有绝对优势;SMARTdenovo相比于其他流程占用资源量较少,且组装结果较为优秀;Canu&PBcR-Self在使用PacBio数据的时候提供了非混合流程中最高的准确率且在数据量减少的情况下依旧能组装出不俗的结果。
数据平台差异分析
两平台数据的组装结果在除了InDel平均数量和线粒体的组装完成度方面以外都表现出了相当高的一致性,因此作者在InDel方面进行了深入地研究。
研究发现,ONT平台在同聚体(homomer)方面的测序结果不如人意,在Canu的组装结果和参考基因组的比对中发现绝大部分的插入缺失都是5-homomers(“AAAAA”,”TTTTT”,”CCCCC”,”GGGGG”),但是有趣的是在用Nanopolish对Canu的组装结果进行修正之后,这些缺失大部分都会被填上,偶尔还会多出一些。
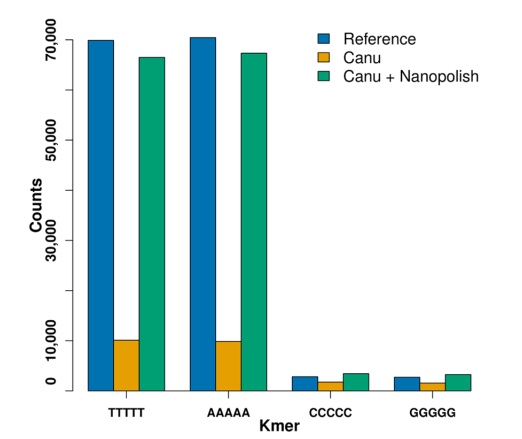
可惜Nanopolish在对于12.1Mb大小基因组的31X2D数据进行处理的时候占用了1835个小时的CPU时间,这也决定了在更大数据量的基因组组装的过程中Nanopolish很难在派上用场。
在线粒体组装的过程中,使用PacBio数据的流程Racon, SMARTdenovo和Canu都完成了完整线粒体的组装,Miniasm也重构出了77%的线粒体,但是在ONT数据方面除了可以使用二代数据进行矫正的PBcR-Miseq流程完成了96%的线粒体基因组组装,其他流程中组装最好的流程Falcon&Canu分别组装了67%和64%的线粒体基因组。
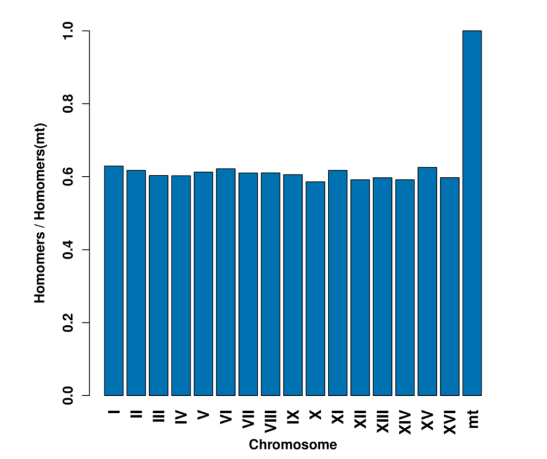
为了查明线粒体组装和核染色体差异的问题,作者将S288C参考基因组每个染色体中的5-homomers按照长度平均后与线粒体的平均5-homomers做比值后发现,线粒体中的5-homomers的比例远高于其他染色体。
总结篇
31X的三代数据就可以组装出可信度高于98%(ONT)、99%(PacBio),Na50s大于550Kb的组装结果,当混入二代数据作为辅助的时候可将可信度提高到99.98%
PacBio的测序数据平均长度明显低于ONT,但是ONT的数据产出量却远低于PacBio,这可能是由于使用的ONT试剂不够稳定,近期的ONT试剂已经表现出了很高的稳定性。同时本文对比的是PacBio RSⅡ和MinION,将一个台式平台与移动平台进行对比无法说明测序方法之间的差异(移动平台总会牺牲部分性能)。
PacBio的测序错误绝大部分为随机错误可以通过加大测序深度来规避该类错误,ONT的测序错误则可能会对某些特殊序列结构如5-homomers有一定程度的偏好性,这种偏好性可能会导致高同聚体比例的线粒体等组装远低于预期。因此ONT的polish步骤很重要,但过于耗时。
在数据组装方面:
a) 二代数据与三代数据的混合流程能组装出可信度最高的组装结果,但是其组装出来的小片段也最多。
b) PBcR-Self和Canu组装出了最连续又精确的基因组,并在三代数据减少的过程中依旧表现出最好的组装能力。















 2839
2839











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









