






 本文详细介绍了Kohn-Sham方程在密度泛函理论中的作用,以及如何通过局域密度近似(LDA)和广义梯度近似(GGA)来估算交换关联泛函。讨论了LDA在计算晶格常数、吸附能等方面的不足,并对比了GGA的改进。最后,概述了求解Kohn-Sham方程的自恰迭代法。
本文详细介绍了Kohn-Sham方程在密度泛函理论中的作用,以及如何通过局域密度近似(LDA)和广义梯度近似(GGA)来估算交换关联泛函。讨论了LDA在计算晶格常数、吸附能等方面的不足,并对比了GGA的改进。最后,概述了求解Kohn-Sham方程的自恰迭代法。
分子模拟学习,请点击蓝字关注~
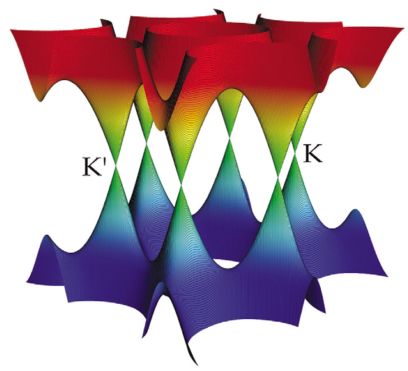
(石墨烯的价带与导带,Materialstoday, Vol 10, 1-2, 2007)
在密度泛函理论的第一篇文章中,我们介绍了Kohn-Sham将多电子体系中的能量进行分解重组,最终建立了能量泛函。除了非关联动能项,核势能,Hartree能,交换关联能都表达成了电子密度的函数,而非关联动能项表达为非关联轨道波函数的泛函,如下式所示:
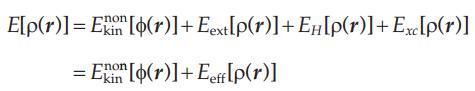
其中各项的表达式为:

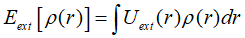
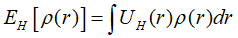
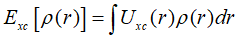
 Kohn-Sham方程
Kohn-Sham方程
下面我们需要求解能量泛函的最小值,也就是体系的基态,在求解能量最小值的时候,需要注意到系统的波函数需要满足正交关系,因此该极值为条件极值。正交限制条件为:
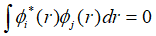
利用拉格朗日乘子法求解极值:
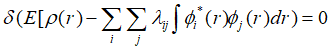
展开上式,代入各项的变分:
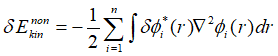
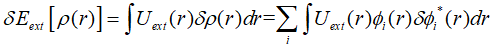
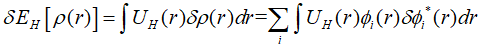
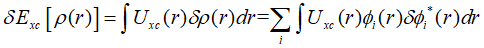
最终,欲使所构造的拉格朗日泛函的变分为0,需要满足:
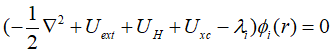
其中拉格朗日乘子是由于各个轨道的正交化而引入的,因此可以看做不同轨道的特征值,也就是各个轨道的特征能量,因此Kohn-Sham方程最终化简为:
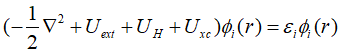
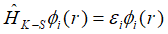
上式就是著名的Kohn-Sham方程,在利用密度泛函理论进行计算的时候,实际的计算都是根据辅助哈密顿量HK-S来进行计算的。
交换关联泛函
利用Kohn-Sham方程求解多电子体系的能量及电子密度的一个重要的前提是:已知的交换关联泛函Uxc。在密度泛函理论中,所有的量在理论上都是精确的,只有交换关联泛函采用的是近似方法得到的。通常的近似方法有局部密度近似(LDA)和广义梯度近似(GGA)。
1. 局域密度近似
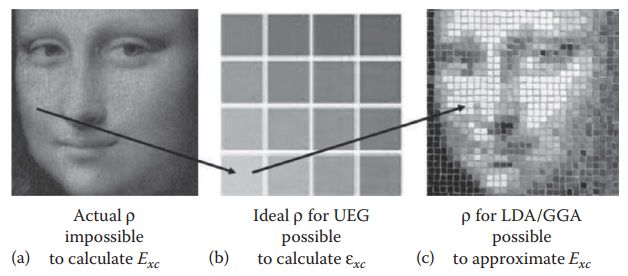
(Book:Computational Materials Science, June Gunn Lee)
局域密度近似的主要思想是,电子在均匀场中将表现为均匀的电子密度,其交换关联能是可以用量子蒙特卡洛算法精确计算的,当外界势场变化幅度不剧烈时,在局域采取均匀的电子密度来计算交换关联能。
在均匀场中,交换能的表达式为:
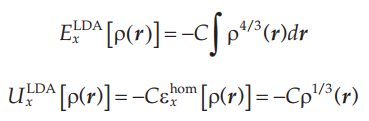
关联能的表达式为:
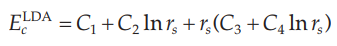
其中Ci都为拟合的常数,rs是与电子密度有关的量,表达式为:
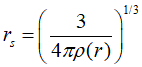
最终交换关联能都表达为电子密度的函数,那么就可以代入Kohn-Sham方程进行计算了。
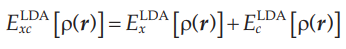
利用局域密度近似也可以处理考虑电子自旋的电子密度:
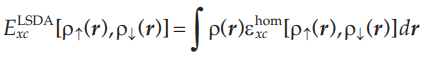
局域密度近似LDA方法在早期的DFT计算中被广泛使用,但它在计算下列问题时存在一定的误差:
1、计算晶格常数时会通常偏小,因此体系的吸附能和体积模量通常偏大。
2、计算吸附能时偏高,计算扩散能垒时偏低。
3、计算电子动量和角动量时偏低。
4、计算能带带宽时,偏低超过50%,有时甚至根本得不到能带带宽。
5、对于过渡态金属的计算不准确,因为这些金属核外电子密度变化剧烈。例如利用LDA计算Fe(铁磁性)和Cr(反铁磁性)两种物质时,最终得到的结果都是非磁性的。
6、对于有氢键和范德华力作用的体系的计算也不精确。
为了获得更精确的交换关联泛函,研究者们提出了广义梯度近似的交换关联泛函。
2. 广义梯度近似
在LDA方法中,交换关联泛函仅为电子密度的函数,而在GGA近似方法中,交换关联泛函是电子密度和电子密度梯度的函数。
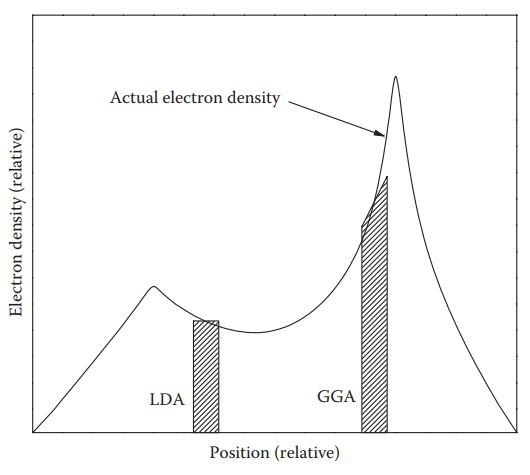
(交换关联泛函LDA和GGA的对比)
GGA形式的交换关联能可以表达为:
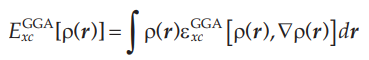
考虑自旋形式的交换关联能的表达式为:
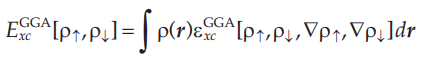
在实际应用中,GGA的表达式其实是直接在LDA的表达式中增加一项与电子密度梯度有关的函数,如下式所示:
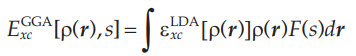
其中:
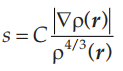
通常s的取值范围为0~3,而F(s)的取值范围为1~1.6。关于F的表达式有许多形式的GGA交换关联泛函,其中比较流行的有PW91和PBE两种类型,分别是Perdew和Wang 1992年和Perdew 1996年提出来的。其中PBE是改进的PW91版本的交换关联泛函,也是目前使用最广泛的,兼顾精度与效率的泛函。下面是利用PBE计算常见材料额晶格常数和体积模量。
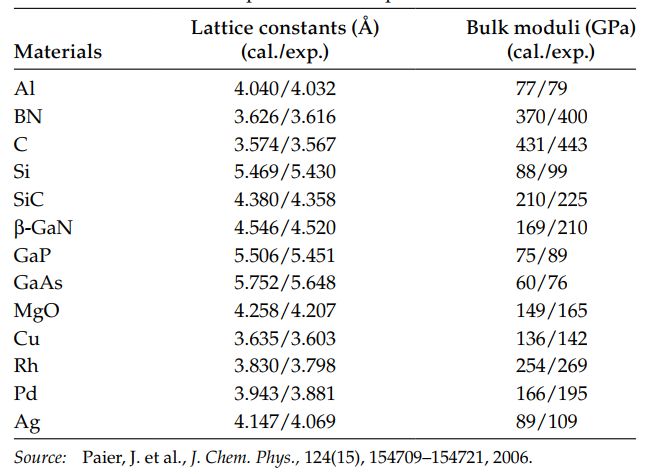
GGA几乎使用于所有的系统,对于结构参数的误差通常为1~3%。但是GGA在计算能带带宽的时候,仍然小于实际值超过50%。造成能带带宽估计偏小的原因有两个:首先,在Kohn-Sham方法中,计算带宽的方法与定义有出入,带宽的定义为:
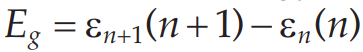
而在Kohn-Sham方法中计算的带宽其实是:
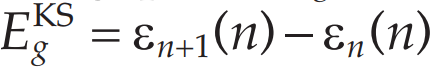
在Kohn-Sham方法中并没有真实的增加或去除电子,所以更高能级n+1轨道的能量也是由n个电子估计的,因此能带带宽偏小。
第二个原因是源于交换关联泛函的连续性,而真实的交换关联泛函应该是不连续的。最终导致同一轨道上相同自旋的电子相互作用不能完全被排除,这就导致了共价带能量的升高,导带能量的降低,所以能带带宽就降低了。在H-F方法中,电子的关联效应可以有Slater行列式完全消除,因此由H-F方法计算的能带带宽通常偏大。
校正能带带宽计算的方法大致有三种:
1、使用混合H-F方法的交换关联泛函,
2、将能量用格林函数表述,利用多体摄动理论校正关联项;
3、使用其他关联项校正的方法。
在实际的DFT计算中,关联密度泛函往往是合并在赝势函数中的,在选择赝势的时候也就选择了交换关联泛函。
Kohn-Sham方程的求解
求解K-S方程通常采用自恰迭代法,如下图所示:
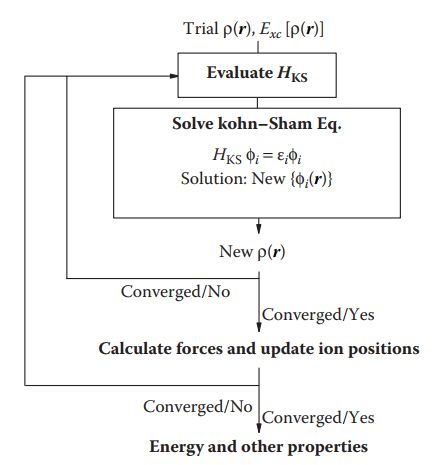
用于初始迭代的波函数是稍微多于n/2个的平面波以及由赝势函数提供的独立的原子周围的电子密度,然后不断代入K-S方程进行迭代,直到能量收敛至基态,达到停止的判断标准,通常为10^-5~10^-4 eV.

分子模拟交流学习,请扫码关注

















 886
886

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









