







本篇是自己学习SAM和SAMtag的资料心得,详细介绍高通量测序中 SAM/BAM格式文件。
本文将了解什么?
欢迎微信搜索关注@pythonic生物人
1、SAM/BAM格式简介 2、术语与概念理解 3、标头部分(header section)详述 4、比对信息部分(alignment section)详述
第一列、QNAME
第二列、FLAG
第三列、RNAME
第四列、POS
第五列、MAPQ
第六列、CIGAR
第七列、RNEXT
第八列、PNEXT
第九列、TLEN
第十列、SEQ
第十一列、QUAL
第十二列之后,Optional fields
1.1 Additional Template and Mapping data(一些比对信息)
1.2 Metadata(这部分内容和 SAM中header section部分相关,描述read测序相关信息)
1.3 Barcodes(UMI/单细胞测序cell barcode)
1.4 Original data
1.5 Annotation and Padding
1.6 Technology-specifific data
2 Locally-defifined tags
1、SAM/BAM格式简介
- SAM存储格式发明的目的:使不同测序平台下机数据,经过不同比对软件后有一个统一的存储格式。
- SAM(Sequence Alignment/Map format简写)格式文件,存储测序数据和参考基因组比对结果的文件,每行以table键分割,包含标头部分(header section)和比对部分(alignment section)见下图。
- BAM(Binary Alignment/Map format简写)格式文件,SAM的二进制格式文件,通过BGZF library参考库压缩而成。

2、术语与概念理解
该部分有助于后文SAM格式理解,后文反复出现如下概念。
模板(Template):一段DNA/RNA序列,它的一部分在测序仪上被测序,或被从原始序列中组装。(意思就是:我们通过测序仪测序的那段序列,或者通过组装原始序列得到的更长的序列,就是模板的一部分)。(从后文来看,对于Illumina双端测序来说,template指的就是插入片段) 片段(Segment):一段连续的序列或子序列(subsequence)(从上下文来看,segment既可以指一条完整的read,也可以指read的一部分); 读段(Read):一段来自测序仪的原始序列。read可以包含多个片段(一条read在比对过程中可能会被拆分成几段,对应到参考序列不同的位置上。read被拆分后形成的片段即为segment)。对于测序数据,reads根据测序顺序进行编号; 线性比对(Linear alignment):一条比对到参考序列上的read可能会有插入、缺失、skips和切除(clipping),但只要没有方向的改变(例如,read的一部分比对到了正义链上,另一部分比对到了反义链上),就是Linear alignment。一个线性比对结果可以代表一个SAM记录;(意思似乎是:一条SAM记录能且只能保存一个线性比对结果) 嵌合比对(Chimeric alignment):不是线性比对的比对。嵌合比对中包含了一套没有大范围重叠的线性比对(嵌合比对中的每一个片段都是线性比对。关于大范围重叠的说法是为了和多重比对区分)。一般地,嵌合比对中的一个线性比对被认为是“有代表性的比对”(representative alignment),而其他的线性比对被称为补充的(supplementary),用补充比对标志(supplementary alignment flag)加以区别(representative和supplementary成一对,对应嵌合比对)。嵌合比对的所有SAM记录有相同的QNAME,其flag值的0x40和0x80位都相同(见1.4节)(0x40位和0x80位分别表示模板中的第一个片段和最后一个片段,为什么会都相同呢?总要有一个是第一个片段,总要有一个是最后一个片段吧,它俩的0x40位和0x80位不应该相同啊?)。哪个线性比对被视为有代表性是任意选择的。(可见嵌合比对中,各个segments的独立性更强:都不在双链的同一条链了。另外,如果一条read的不同部分比对到了不同的染色体上,那肯定也是嵌合比对了,因为不同染色体之间讨论方向相同是没有意义的,肯定不可能是线性比对了。) read比对(read alignment):能代表一条read的比对结果的线性比对或嵌合比对; 多重比对(Multiple mapping):由于重复序列等情况的存在,一条read在参考基因组上的正确位置可能无法确定。在这种情况下,一条read可能会有多种比对结果,其中一种被视为主要的(primary),所有其他的比对结果的SAM记录的flag标志中都会有一个“次要(secondary)比对结果”的标志。所有这些SAM记录拥有相同的QNAME,flag标志的0x40位和0x80位有相同的值。一般被指定为“主要”的比对结果是最佳比对,如果都是最佳比对,则任意指定一条(primary和secondary成一对,对应多重比对)。(原文注释:嵌合比对主要由结构变异、基因融合、组装错误、RNA测序或实验过程中的一些原因造成,更经常出现在长reads中(长read有利于检测嵌合比对。这就是为什么三代测序是检测染色体结构变异的更有力工具)。嵌合比对中的线性比对之间没有大片段的重叠,每个线性比对有较高的mapping质量值,可以用于SNP/INDEL的检测;而多重比对主要是序列重复造成的,不经常出现在长reads中。如果一条read有多重比对的情况,所有的比对互相之间几乎完全完全重叠。除了一个最佳比对外,所有其他比对的质量值都<3,且会被大多数SNP/INDEL检测软件忽略)。 以1为起始的坐标系(1-based coordinate system):序列的第一位是1的坐标系。在这种坐标系中,一个区域用闭区间表示。例如,第三位和第七位碱基之间的区域表示为[3,7]。SAM, VCF, GFF和Wiggle格式使用以1为起始的坐标系; 以0为起始的坐标系(0-based coordinate system):序列的第一位是0的坐标系。在这种坐标系中,一个区域用左闭右开区间表示。例如,第三位和第七位碱基之间的区域表示为[2,7)(原文如此。难道不应该是[3,8)么?不应该。以0为起始,第三位对应的索引号是2,第七位对应的索引号是6,所以索引号[2,7)对应了第三位-第七位碱基。当时脑子糊涂了,没搞清文中说的意思)。BAM, BCFv2, BED和PSL格式使用以0为起始的坐标系; Phred scale:如果一个概率值0<p≤1,这个p值的phred scale等于-10log(10)p,舍入为最近的整数。
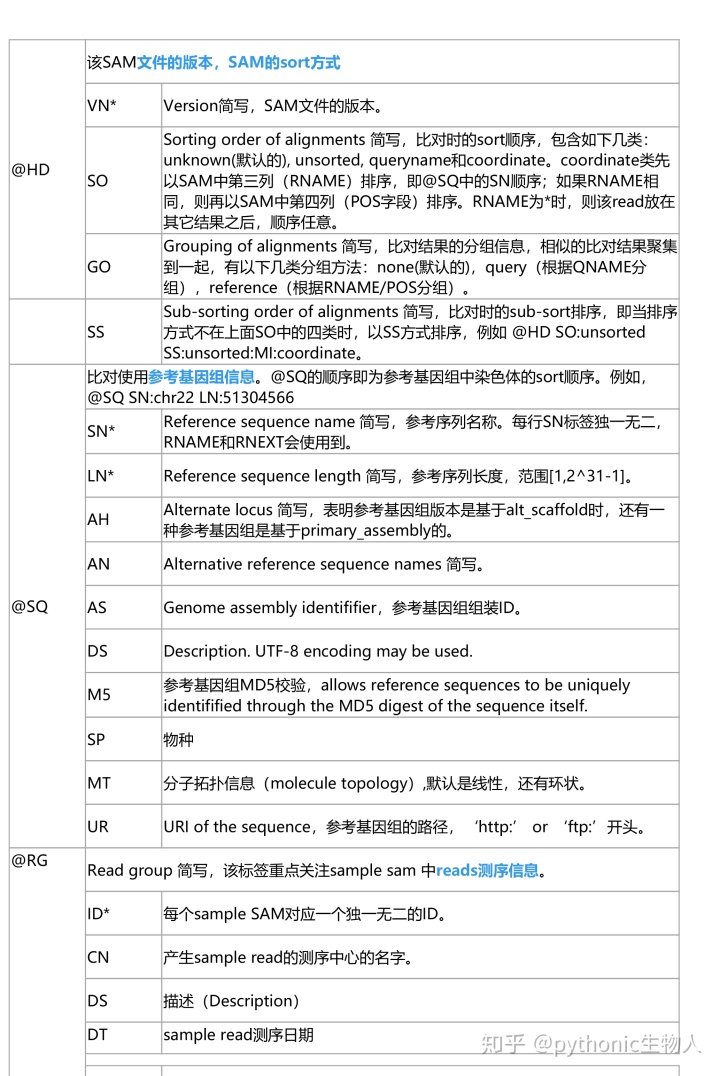
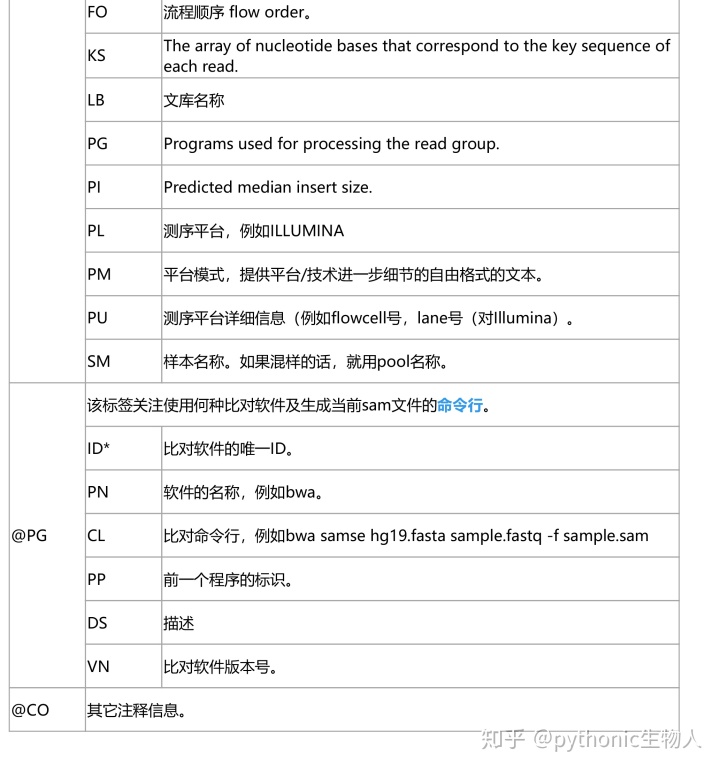
3、标头部分(header section)详解

该部分为SAM/BAM的注释部分,该部分并非必须,可以省略。每一行都以@符开头,后面跟着两个大写字母,每个字段之间以t分割,每个字段遵循(TAG:Value)的格式(@CO开头的行除外)。每行可以使用以下正则表达式表示:/^@(HD|SQ|RG|PG)(t[A-Za-z][A-Za-z0-9]:[ -~]+)+$/ or /^@COt.*/,@后紧跟的两个大写字母主要有HD,SQ,RG,PG和CO五类,前四类常用如下表,其中加了*号的表示该标签必须存在,例如@HD这个标签存在时,VN必须同时存在,详细介绍如下。
4、比对信息部分(alignment section)详解
- 比对部分概述
该部分是SAM文件的核心部分,每一行代表一个序列的线性比对(linear alignment of a segment),每行包含前11个必需字段,和第
 最低0.47元/天 解锁文章
最低0.47元/天 解锁文章
















 5596
5596











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









