






通常我们在挖掘TCGA数据库的时候,会发现该项目纳入的正常组织测序结果是非常少的,也就是说很多病人都不会有他的正常组织的转录组测序结果,比如说乳腺癌吧,1200个左右的转录组数据,其中1100左右都是肿瘤组织的测序数据,只有区区100个左右的正常对照。
这个时候我们就需要想办法加大正常组织测序样本量,既然TCGA数据库没有,我们就从其他数据库着手。
背景知识
一期
2015年,GTEx发布了第一个阶段性成果,一次性在Science杂志上发表三篇研究成果,该成果还被选为封面文章。GTEx的研究从175名死者身上采集到了1641个尸检样本,这些样本来自54个不同的身体部位,对几乎所有转录基因的基因表达模式进行了观察,从而够确定基因组中影响基因表达的特定区域。另外两篇文章之一从人所有组织中的基因表达谱进行了描述,证明了组织特异性的某些基因往往决定了组织特异性基因的表达调控;另一篇解释了截短的蛋白变异体如何影响组织中的基因表达。
二期
在2017年,一次性在nature发表4篇研究成果,GTEx研究联盟的研究收集并研究了来自449名生前健康的人类捐献者的7000多份尸检样本,涵盖44个组织(42种不同的组织类型),包括31个实体器官组织、10个脑分区、全血、两个来自捐献者血液和皮肤的细胞系,作者利用这些样本研究基因表达在不同组织和个体中有何差异。题为“Landscape of X chromosome inactivation across human tissues”和“Dynamic landscape and regulation of RNA editing in mammals”的论文,采用GTEx数据探讨了与基因表达相关联的基因变异如何能够调节RNA编辑和X染色体失活现象。
数据库内容介绍
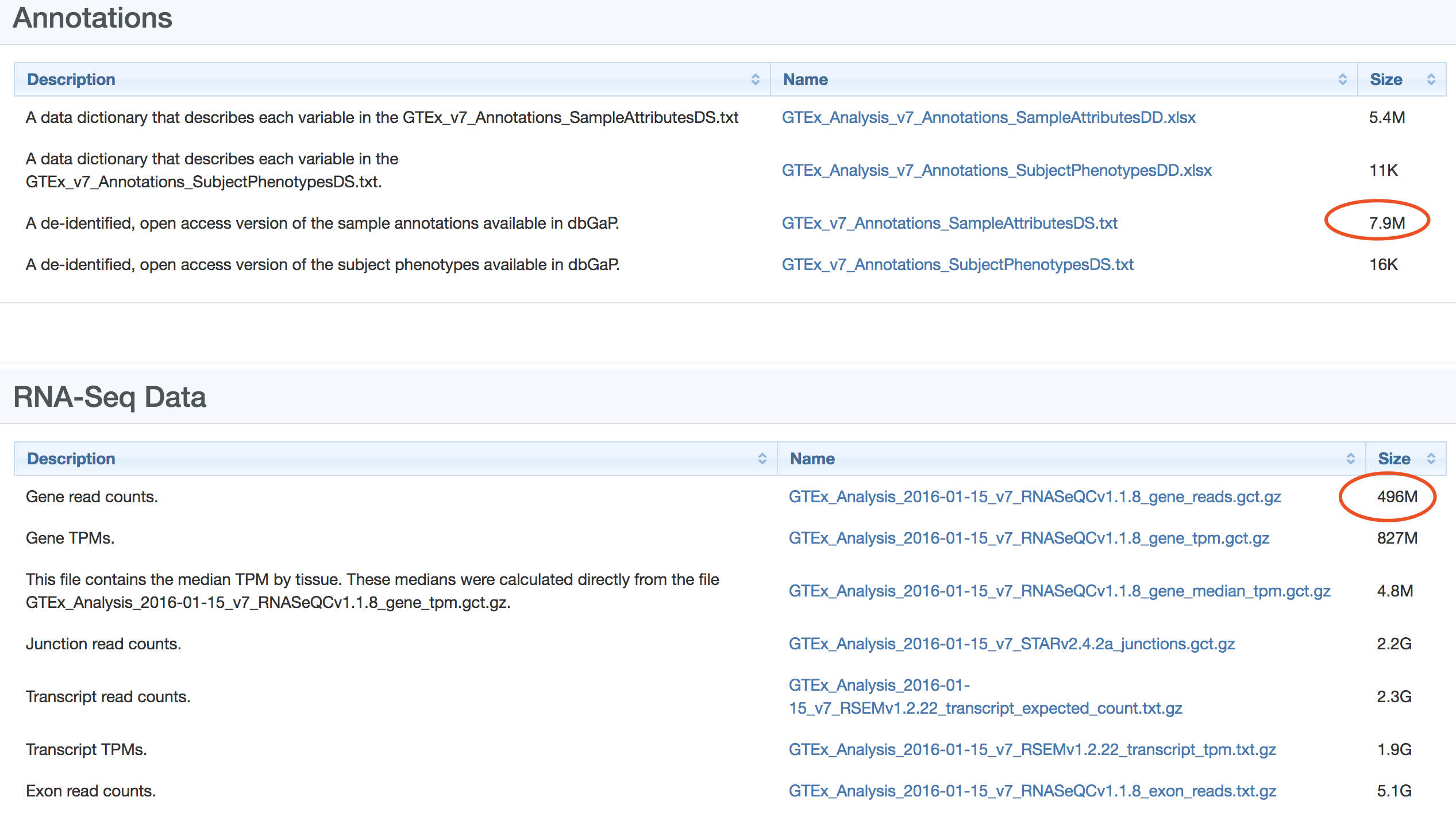
其中,对我们来说最重要的就是 表达矩阵, 可以下载图中 gene read counts 这个496M的文件,表达矩阵里面的样本ID肯定是数据库组织者自定义的,所以我们还需要找到样本ID的注释信息。
更多的是关于这个数据库的网页使用介绍,我们生信工程师通常不需要,就不赘述了。
注意一下 数据库的版本信息:
The current release is V7 including 11,688 samples, 53 tissues and 714 donors
首先看数据库的注释信息
重点是:
# SMTS Tissue Type, area from which the tissue sample was taken.
# SMTSD Tissue Type, more specific detail of tissue type
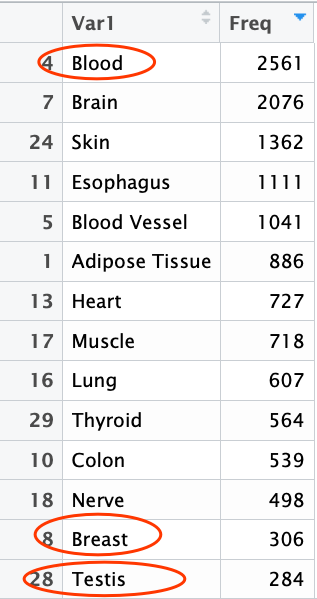
可以看到每个样本属于哪一种组织,这样就方便提取他们的信息来辅助自己的研究。
把 gene read counts 这个496M的表达矩阵导入R:
if(F){
options(stringsAsFactors = F)
GTEx=read.table('~/Downloads/GTEx_Analysis_2016-01-15_v7_RNASeQCv1.1.8_gene_reads.gct.gz'
,header = T,sep = '\t',skip = 2)
GTEx[1:4,1:4]
h=head(GTEx)
save(h,file = 'GTEx_head.Rdata')
}
挑选感兴趣的组织的表达矩阵
上面我们详细了解了不同样本注释到的组织,所以代码很简单:
load('~/Desktop/GTEx_all.Rdata')
a[1:4,1:4]
colnames(a)
# SMTS Tissue Type, area from which the tissue sample was taken.
# SMTSD Tissue Type, more specific detail of tissue type
b=read.table('GTEx_v7_Annotations_SampleAttributesDS.txt',
header = T,sep = '\t',quote = '')
table(b$SMTS)
breat_gtex=a[,gsub('[.]','-',colnames(a)) %in% b[b$SMTS=='Breast',1]]
rownames(breat_gtex)=a[,1]
dat=breat_gtex
就是把属于breast这个组织的样本名挑选出来,在上面的表达矩阵里面取子集即可。
值得注意的是这个时候的表达矩阵基因名不是symbol,是需要进行ID转换的,代码如下:
dat=breat_gtex
ids=a[,1:2]
head(ids)
colnames(ids)=c('probe_id','symbol')
dat=dat[ids$probe_id,]
dat[1:4,1:4]
ids$median=apply(dat,1,median)
ids=ids[order(ids$symbol,ids$median,decreasing = T),]
ids=ids[!duplicated(ids$symbol),]
dat=dat[ids$probe_id,]
rownames(dat)=ids$symbol
dat[1:4,1:4]
breat_gtex=dat
save(breat_gtex,file = 'breat_gtex_counts.Rdata')
表达矩阵如下所示:

正常乳腺组织样本表达矩阵可以进行的分析
通常情况下应该是去和肿瘤数据进行分析,那样的分析就多元化了,这里来个简单点的,可以进行pam50分类:
if(T){
ddata=t(dat)
ddata[1:4,1:4]
s=colnames(ddata);head(s)
library(org.Hs.eg.db)
s2g=toTable(org.Hs.egSYMBOL)
g=s2g[match(s,s2g$symbol),1];head(g)
# probe Gene.symbol Gene.ID
dannot=data.frame(probe=s,
"Gene.Symbol" =s,
"EntrezGene.ID"=g)
ddata=ddata[,!is.na(dannot$EntrezGene.ID)]
dannot=dannot[!is.na(dannot$EntrezGene.ID),]
head(dannot)
library(genefu)
# c("scmgene", "scmod1", "scmod2","pam50", "ssp2006", "ssp2003", "intClust", "AIMS","claudinLow")
s
annot=dannot,do.mapping=TRUE)
table(s$subtype)
tmp=as.data.frame(s$subtype)
subtypes=as.character(s$subtype)
}
library(genefu)
pam50genes=pam50$centroids.map[c(1,3)]
pam50genes[pam50genes$probe=='CDCA1',1]='NUF2'
pam50genes[pam50genes$probe=='KNTC2',1]='NDC80'
pam50genes[pam50genes$probe=='ORC6L',1]='ORC6'
x=dat
x=x[pam50genes$probe[pam50genes$probe %in% rownames(x)] ,]
tmp=data.frame(subtypes=subtypes)
rownames(tmp)=colnames(x)
library(pheatmap)
pheatmap(x,show_rownames = T,show_colnames = F,
annotation_col = tmp,
filename = 'ht_by_pam50_raw.png')
x=t(scale(t(x)))
x[x>1.6]=1.6
x[x< -1.6]= -1.6
pheatmap(x,show_rownames = T,show_colnames = F,
annotation_col = tmp,
filename = 'ht_by_pam50_scale.png')
单独取出pam50包含的50个基因的表达矩阵进行热图聚类:
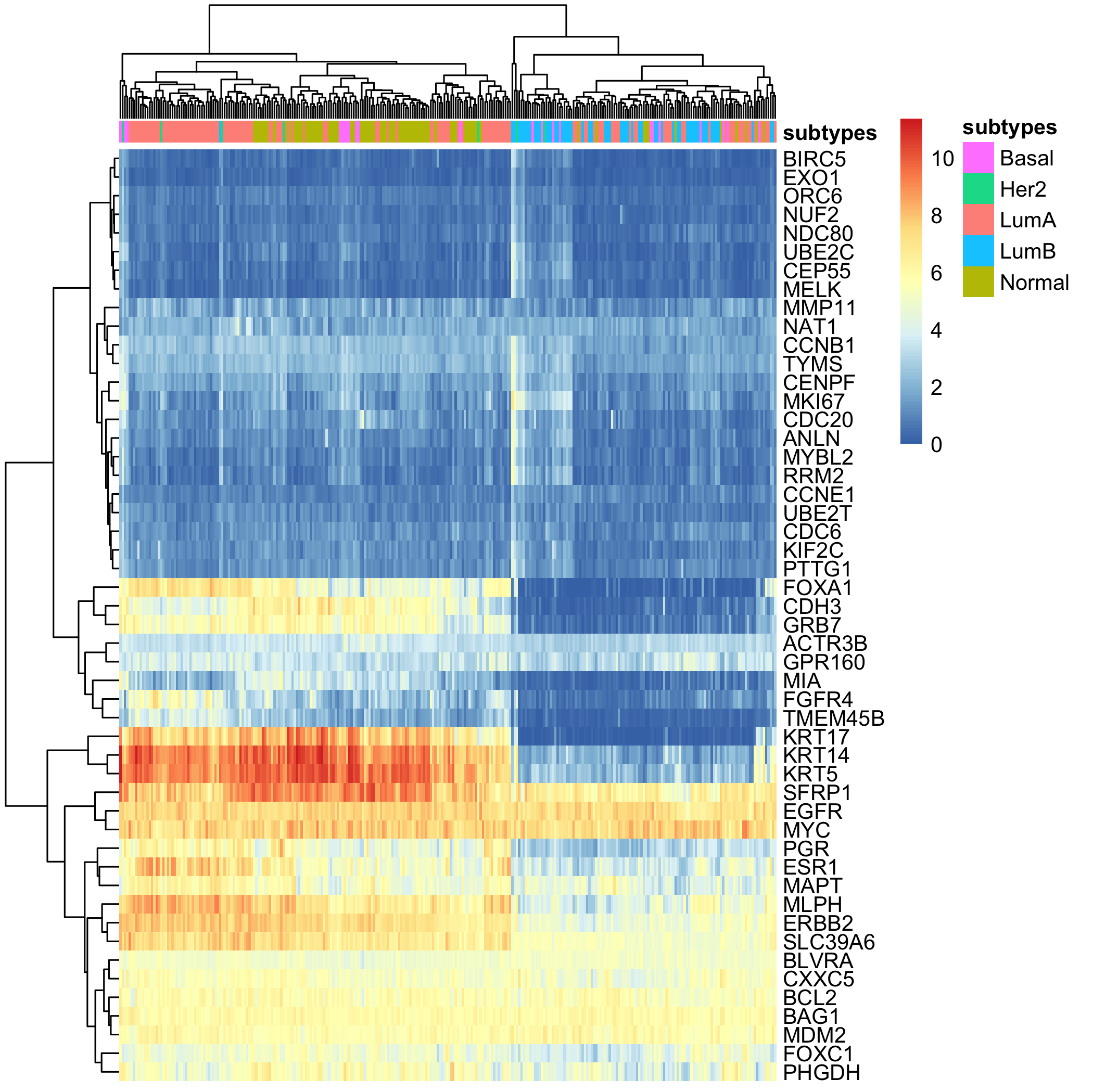
由上图可以看到不同基因的表达量是 差异很大的,通常我们不会去比较不同基因的表达量,而只是比较同一个基因在不同样本的表达量差异的。
所以我们没有必要去看不同基因的表达量高低,那么就可以进行一定程度的归一化,重新绘图如下:

可以很明显的看到哪怕是对正常组织的转录组测序结果走pam50的分类也是可以拿到各种各样的分类结果的。
但是pam50的分类是在乳腺癌患者的芯片表达矩阵进行训练的模型,是因为我们用错了地方,可以看看在METEBRIC里面的分类结果:
上面的分类是pam50算法的结果,下面的分类是临床信息。
可以看到basal的结果还是很统一的,而且都比较符合TNBC的定义,就是PGR,ESR1,ERBB2都表达量都很低。

去除批次效应
如果真的要把GTEx数据库的转录组表达矩阵和TCGA的进行比较,还需要一定程度的去除批次效应。
我以前在生信技能树多次讲解,这里也不再赘述。















 6840
6840











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









