






Title
题目
Joint modeling histology and molecular markers for cancer classification
联合建模组织学和分子标记用于癌症分类
01
文献速递介绍
癌症是对人类致命的恶性肿瘤,早期准确诊断对癌症治疗至关重要。目前,病理诊断仍是癌症诊断的金标准,但传统病理诊断劳动强度大、耗时长,且高度依赖神经病理学家的专业经验。数字病理学通过自动化算法分析组织全玻片图像(WSIs)(Lu等人,2021),有望实现快速诊断,为及时精准治疗提供可能。 近年来,基于深度学习(DL)的数字病理学方法已成功应用于多种癌症的诊断(Jose等人,2023)。这些方法主要基于病理诊断标准,利用组织学特征进行癌症诊断。与此同时,过去十年中,癌症诊断分子标记的发现激增,推动了癌症诊断标准从传统组织病理学向分子病理学的范式转变。例如,胶质瘤是成人中最常见的恶性原发性肿瘤之一,高级别胶质瘤的中位总生存期不足14个月(Molinaro等人,2019;Zhang等人,2024b)。2021年世界卫生组织(WHO)脑肿瘤分类(Louis等人,2021)确立了几个关键分子标记,如异柠檬酸脱氢酶(IDH)突变、1p/19q染色体共缺失,以及细胞周期蛋白依赖性激酶抑制剂2A/B(CDKN)纯合缺失(HOMDEL)。根据更新的标准,胶质母细胞瘤主要基于IDH突变进行诊断,而此前纯粹基于坏死和微血管增殖(NMP)等组织学特征。此外,IDH和CDKN状态已定义了一类新的高级别星形细胞瘤。这一显著的范式转变革新了癌症的临床路径,也为开发新型数字病理学方法提供了新机遇。 将分子标记与组织学整合到临床诊断中仍面临若干实际挑战:首先,基因测序和免疫染色等分子标记评估方法通常耗时且成本高昂;其次,组织学评估与分子标记遵循独立的诊断流程,阻碍了临床诊断中两者的及时有效整合。深度学习方法有望通过独特优势解决上述挑战:(1)从全玻片图像预测分子标记:越来越多研究表明,组织学特征与分子改变相关,使得直接从全玻片图像预测分子标记成为可能(Wang等人,2024a);(2)整合诊断流程:基于深度学习的方法可有效整合分子标记与组织学特征的诊断流程,并对不同数据类型间的相互作用进行建模,从而推动最新诊断范式下的癌症诊断。因此,迫切需要开发能够联合预测分子标记和组织学特征并捕捉其相互作用的深度学习方法,这一整合方法有望简化并提高癌症诊断的准确性。 本文提出一种新型数字病理学方法——癌症分类多尺度多任务建模(M3C2),以适应新兴的分子病理学癌症诊断范式。既往研究尝试整合组织学与基因组学用于肿瘤诊断(Xing等人,2022),例如Ding等人(2023)开发了一种基于无监督预训练的多模态Transformer,整合病理与基因组学以预测结肠癌生存。尽管取得了一定成功,但现有方法大多仅将分子标记作为补充输入,无法同时预测组织学和分子标记并进一步建模其相互作用,这限制了其在当前诊断方案中的临床适用性。为解决这一局限并匹配更新的临床诊断路径,我们利用基于视觉Transformer(Dosovitskiy等人,2020)的新型分层多任务框架,通过两个部分权重共享的组件联合预测组织学和分子标记,有效建模其相互作用并增强临床相关性。 首先,模型设计为从全玻片图像的多个放大倍数中提取有效特征。在临床实践中,病理诊断涉及在不同放大倍数下观察组织切片,例如从20倍(细胞水平,0.25 μm/像素)到10倍(组织水平,1 μm/像素)(Schmitz等人,2021)。因此,我们设计了多尺度解缠模块以捕捉癌症诊断所需的关键全玻片图像特征,尤其通过新型解缠损失高效提取适用于组织学和分子标记预测的特征。 其次,聚焦于建模不同分子标记间的关系。由于潜在的癌症进化和生物学机制,分子标记之间存在内在关联,因此指南通常需要多个分子标记以实现准确癌症分类。为模拟真实场景,我们将多个分子标记的预测构建为多标签分类(MLC)任务。尽管既往多标签分类方法有效捕捉了标签相关性(Li等人,2022b),但现有方法可能忽略预测过程中分子标记的共现性和内在关联(Zhang等人,2023)。为此,我们提出基于共现概率的标签关联图(CPLC-Graph)网络,以建模分子标记的共现性和相互关系。 最后,对分子标记与组织学预测间的相互作用进行建模。具体而言,引入跨模态交互模块以捕捉分子标记与组织学特征(如IDH突变与NMP,均为胶质母细胞瘤诊断的关键特征)之间的相互作用。在此模块中,首先设计动态置信度约束(DCC)损失,引导模型聚焦于两项任务(组织学与分子标记预测)的相似全玻片图像区域;除损失层面的交互外,还开发了跨模态梯度调制(CMG-Modu)学习策略,以协调组织学与分子标记预测的训练过程。据我们所知,这是首次通过建模组织学与分子标记预测的相互作用进行癌症分类的尝试。 本文主要贡献如下:(1)提出多任务多实例学习框架,联合预测组织学和分子标记并对胶质瘤进行分类,反映了最新诊断范式;(2)设计多尺度解缠模块,为组织学和分子标记预测生成高效多尺度特征,并提出CPLC-Graph网络建模多个分子标记间的关系;(3)设计DCC损失和CMG-Modu训练策略,协调组织学与分子标记间的跨模态交互以用于胶质瘤分类;(4)通过广泛实验验证了模型在胶质瘤分类、分子标记和组织学预测等多项任务中的性能。
Abatract
摘要
Cancers are characterized by remarkable heterogeneity and diverse prognosis. Accurate cancer classification isessential for patient stratification and clinical decision-making. Although digital pathology has been advancingcancer diagnosis and prognosis, the paradigm in cancer pathology has shifted from purely relying on histologyfeatures to incorporating molecular markers. There is an urgent need for digital pathology methods to meetthe needs of the new paradigm. We introduce a novel digital pathology approach to jointly predict molecularmarkers and histology features and model their interactions for cancer classification. Firstly, to mitigate thechallenge of cross-magnification information propagation, we propose a multi-scale disentangling module,enabling the extraction of multi-scale features from high-magnification (cellular-level) to low-magnification(tissue-level) whole slide images. Further, based on the multi-scale features, we propose an attention-basedhierarchical multi-task multi-instance learning framework to simultaneously predict histology and molecularmarkers. Moreover, we propose a co-occurrence probability-based label correlation graph network to modelthe co-occurrence of molecular markers. Lastly, we design a cross-modal interaction module with the dynamicconfidence constrain loss and a cross-modal gradient modulation strategy, to model the interactions of histologyand molecular markers. Our experiments demonstrate that our method outperforms other state-of-the-artmethods in classifying glioma, histology features and molecular markers. Our method promises to promoteprecise oncology with the potential to advance biomedical research and clinical applications. The code isavailable at github.
癌症具有显著的异质性和多样的预后特征,准确的癌症分类对于患者分层和临床决策至关重要。尽管数字病理学已推动癌症诊断和预后评估的发展,但癌症病理学的范式已从单纯依赖组织学特征转向整合分子标记。目前迫切需要满足这一新范式需求的数字病理学方法。为此,我们提出一种新型数字病理学方法,通过联合预测分子标记与组织学特征并建模其相互作用以实现癌症分类。 ### 方法核心创新 1. 多尺度解缠模块 为解决跨放大倍数的信息传播挑战,设计了从高倍(细胞水平)到低倍(组织水平)全玻片图像中提取多尺度特征的模块,实现不同层级病理特征的有效分离与融合。 2. 基于注意力的分层多任务多实例学习框架 在多尺度特征基础上,构建分层框架以同时预测组织学特征和分子标记。通过注意力机制聚焦关键区域,并利用多实例学习处理全玻片图像的海量数据特性,提升预测精度。 3. 基于共现概率的标签关联图网络 设计图网络建模分子标记的共现关系,通过共现概率矩阵捕捉标记间的依赖模式,揭示癌症分子特征的内在关联。 4. 跨模态交互模块与动态约束策略 引入带动态置信度约束损失的跨模态交互模块,结合跨模态梯度调制策略,强化组织学与分子标记特征的交互建模,确保信息互补性的有效利用。 实验验证与性能 在胶质瘤分类任务中,所提方法在组织学特征和分子标记分类中均优于现有先进方法,验证了联合建模策略的有效性。该研究为精准肿瘤学提供了新工具,有望推动生物医学研究和临床应用的发展。 代码开源 相关代码已在GitHub平台开源,供学术界和工业界参考使用。
Method
方法
3.1. Framework
According to the latest glioma diagnosis criteria using both histology and molecular information, it is therefore intuitive to jointly learnthe multiple tasks of histology and molecular markers prediction, aswell as the final glioma classification, in a unified framework. In thispaper, we propose a novel M3C2 method to simultaneously handlethese tasks. The framework of M3C2 is shown in Fig. 1. As can beseen, given the cropped multi-scale patches {𝐗 ℎ 𝑖 } 𝑁 𝑖=1 and {𝐗 𝑙 𝑗 } 𝑁 𝑗=1 ∈R𝑁×𝐻×𝑊 ×3 (with patch number 𝑁, height 𝐻, width 𝑊 and 3 channelsof RGB) of 20X and 10X WSI magnification as the input, the modelcan predict (1) molecular markers, including IDH mutation 𝑙 ̂ 𝑖𝑑ℎ ∈ R2 ,1p/19q co-deletion 𝑙 ̂ 1𝑝∕19𝑞 ∈ R2 and CDKN HOMDEL 𝑙 ̂ 𝑐𝑑𝑘𝑛 ∈ R2 , (2)existence of NMP 𝑙 ̂ 𝑛𝑚𝑝 ∈ R2 and (3) final diagnosis of glioma 𝑙 ̂ 𝑔𝑙𝑖𝑜 ∈ R4 .Note that in our 4-class glioma classification task, class 0 to 3 referto grade 4 GBM, high grade astrocytoma, low grade astrocytoma andoligodendroglioma, respectively.The structure of M3C2 consists of 4 modules, including multi-scaledisentangling module, molecular prediction module, histology prediction module and cross-modal interaction module. Detailed structuresare described as follows.
3.1 框架 根据最新的结合组织学和分子信息的胶质瘤诊断标准,在统一框架中联合学习组织学特征预测、分子标记预测及最终胶质瘤分类等多项任务是直观可行的。本文提出一种新型M3C2方法以同步处理这些任务,其框架如图1所示。如图所示,模型以20倍和10倍放大倍数全玻片图像(WSI)的裁剪多尺度图像块{𝐗ₕᵢ}ₙᵢ=1和{𝐗ₗⱼ}ₙⱼ=1∈Rᴺ×ᴴ×ᵂ×3(图像块数量为N,高H、宽W,3通道RGB)为输入,可预测:(1)分子标记,包括IDH突变𝑙̂ᵢᵈʰ∈R²、1p/19q共缺失𝑙̂₁ₚ∕₁₉q∈R²和CDKN纯合缺失𝑙̂cdkn∈R²;(2)坏死和微血管增殖(NMP)的存在性𝑙̂nmp∈R²;(3)胶质瘤最终诊断𝑙̂glio∈R⁴。需要说明的是,在我们的4类胶质瘤分类任务中,0到3类分别对应4级胶质母细胞瘤(GBM)、高级别星形细胞瘤、低级别星形细胞瘤和少突胶质细胞瘤。 M3C2的结构包含4个模块,即多尺度解缠模块、分子标记预测模块、组织学特征预测模块和跨模态交互模块。具体结构描述如下:
Conclusion
结论
The paradigm of pathology diagnosis of diffuse gliomas has shiftedto integrating molecular makers with histology features. In this paper,we target on classifying glioma under the latest diagnosis criteria, viajointly learning the tasks of molecular marker and histology prediction,as well as the final glioma classification. Inputting multi-magnificationhistology WSIs, our model incorporates a novel AHMT-MIL frameworkwith multi-scale disentangling to extract both cellular-level and tissuelevel information for the downstream tasks. Moreover, a CPLC-Graphnetwork is devised for intra-omic interactions, while a DCC loss and aCMG-Modu training strategy are further designed for inter-omic interactions. Our experiments demonstrate that M3C2 achieves superior andmore robust performance over other state-of-the-art methods, openinga new avenue of for digital pathology based on WSIs in the era ofmolecular pathology.
弥漫性胶质瘤的病理诊断范式已转向分子标记与组织学特征整合。本文针对最新诊断标准下的胶质瘤分类,通过联合学习分子标记预测、组织学特征预测及最终胶质瘤分类任务,提出一种新方法。模型以多放大倍数组织学全玻片图像(WSIs)为输入,采用新型分层多任务多实例学习框架(AHMT-MIL)结合多尺度解缠模块,提取细胞级和组织级信息以支持下游任务。此外,设计CPLC-Graph网络建模分子标记间的内在关联(组内交互),并通过动态置信度约束损失(DCC loss)和跨模态梯度调制策略(CMG-Modu)强化分子与组织学特征的跨模态交互(组间交互)。实验表明,M3C2在性能和鲁棒性上均优于现有先进方法,为分子病理时代基于全玻片图像的数字病理学开辟了新路径。
Figure
图
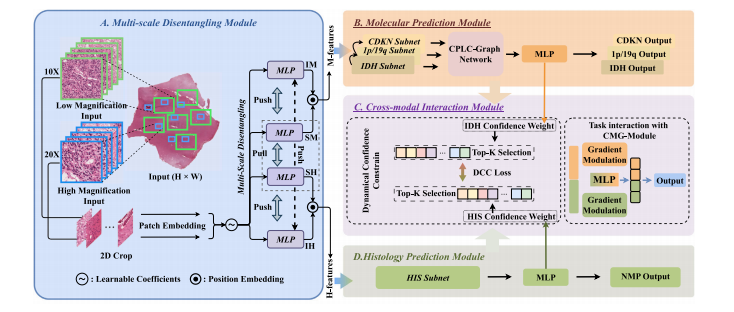
Fig. 1. Framework of our M3C2 method, including (A) multi-scale disentangling module, (B) molecular prediction module, (C) cross-module interaction module and (D) histologyprediction module. Note that IM, SM, IH and SH denote independent molecular features, shared molecular features, independent histology features and shared histology features,respectively
图1. 我们的M3C2方法框架,包括(A)多尺度解缠模块,(B)分子标记预测模块,(C)跨模块交互模块和(D)组织学预测模块。请注意,IM、SM、IH和SH分别表示独立分子特征、共享分子特征、独立组织学特征和共享组织学特征。
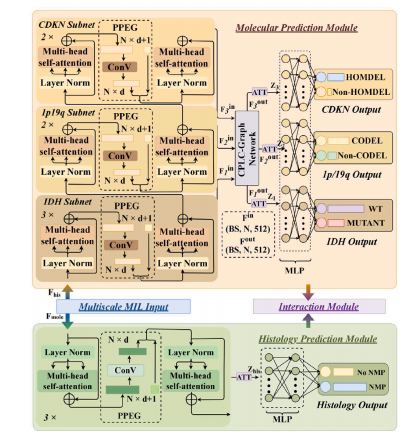
Fig. 2. Detailed structure of the proposed molecular prediction module (above) andthe histology prediction module (below).
图2. 所提出的分子标记预测模块(上)和组织学预测模块(下)的详细结构。
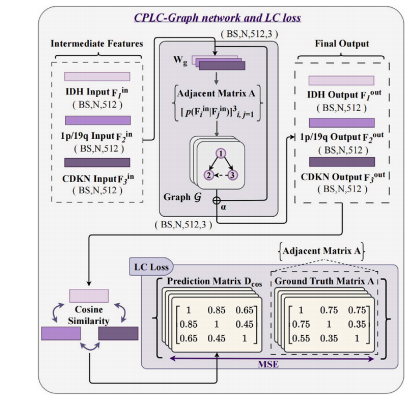
Fig. 3. Detailed structure of the proposed CPLC-Graph network and the LC loss.
图3. 所提出的CPLC-Graph网络及LC损失的详细结构。
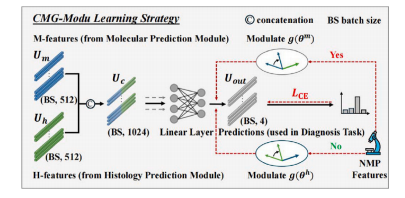
Fig. 4. Illustration of the CMG-Modu learning strategy
图4. CMG-Modu学习策略示意图

Fig. 5. ROCs of our model, comparison and ablation models for predicting IDH, 1p/19q, CDKN, NMP and Glioma.
图5. 本模型、对比模型及消融模型用于预测IDH、1p/19q、CDKN、NMP和胶质瘤的受试者工作特征曲线(ROC)
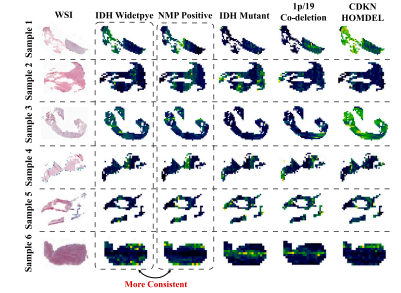
Fig. 6. Visualization maps of M3C2 predicting molecular markers of IDH mutation,1p/19q co-deletion and CDKN HOMDEL, as well as histology of NMP.
图6. M3C2模型预测IDH突变、1p/19q共缺失、CDKN纯合缺失(HOMDEL)分子标记及NMP组织学特征的可视化图
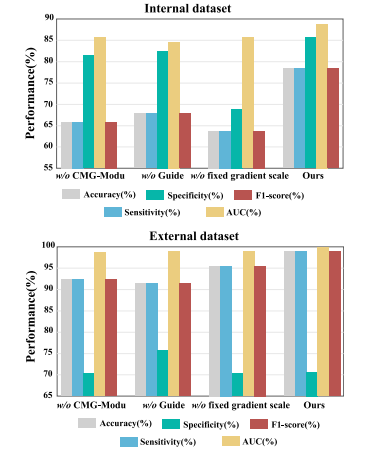
Fig. 7. Ablation study of CMG-Modu training strategy on the glioma classification taskover internal and external validation datasets.
图7. 内部和外部验证数据集上CMG-Modu训练策略对胶质瘤分类任务的消融研究
Table
表
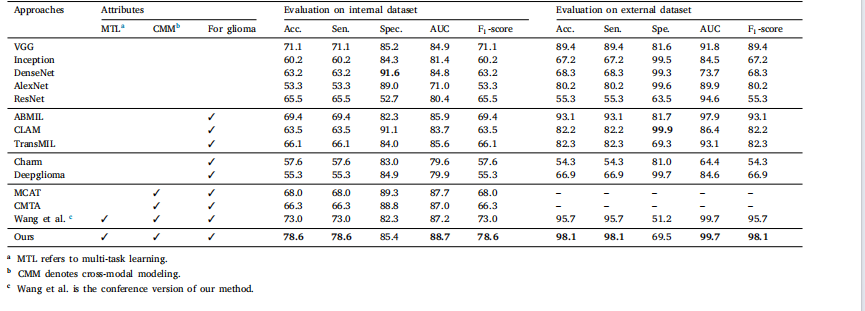
Table 1Mean values in terms of percentage for glioma classification metrics by our and other methods over the internal and external validation datasets
表 1 我们的方法与其他方法在内部和外部验证数据集上的胶质瘤分类指标(百分比平均值)
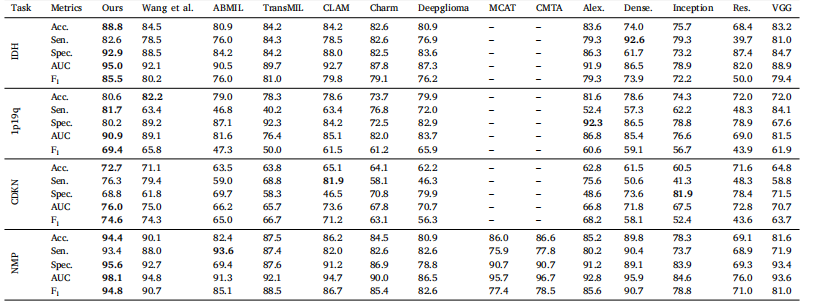
Table 2Mean values in terms of percentage for predicting molecular markers and histology over the internal dataset.
表 2 内部数据集上分子标记和组织学预测的指标平均值(百分比)
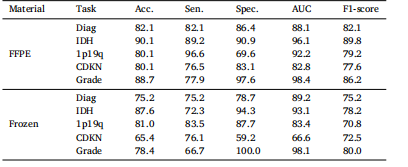
Table 3Subgroup analysis in terms of WSI materials on multiple tasks of molecular markersand histology prediction, as well as glioma classification
表3 基于全玻片图像(WSI)样本类型的分子标记与组织学预测及胶质瘤分类多任务亚组分析
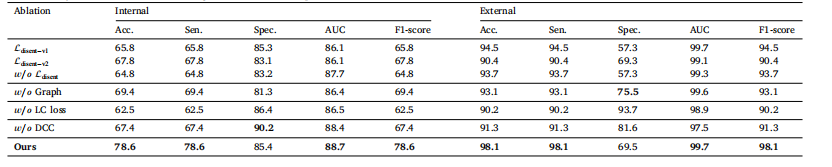
Table 4Ablation study on the multi-modal disentanglement loss, CPLC-Graph network, LC loss and DCC loss on the internal and external validation datasets.
表 4 多模态解缠损失、CPLC-Graph 网络、LC 损失和 DCC 损失在内部和外部验证数据集上的消融研究
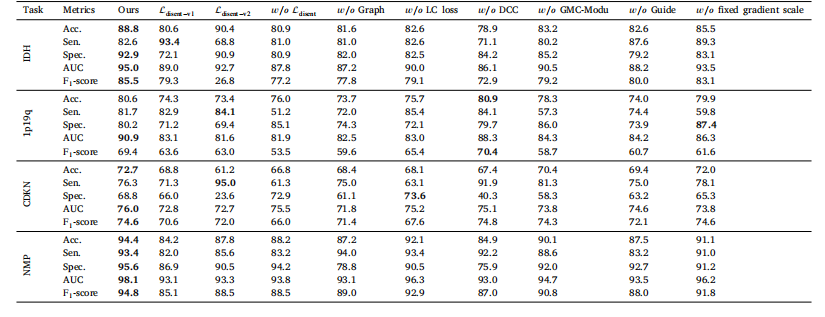
Table 5Ablation study on the multi-modal disentanglement loss, CPLC-Graph network, LC loss, DCC loss and CMG-Modu training strategy on the auxiliary tasks of molecular markers andhistology prediction over internal dataset
表 5 多模态解缠损失、CPLC-Graph 网络、LC 损失、DCC 损失和 CMG-Modu 训练策略在内部数据集分子标记和组织学预测辅助任务上的消融研究
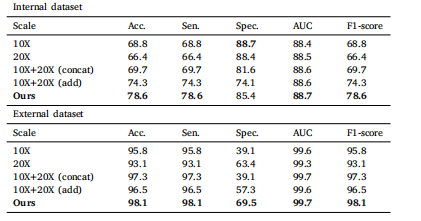
Table 6Mean values in terms of percentage for glioma classification accuracy by our and othermagnification-related baseline methods.
表 6 本方法与其他放大倍数相关基线方法的胶质瘤分类准确率(百分比平均值)
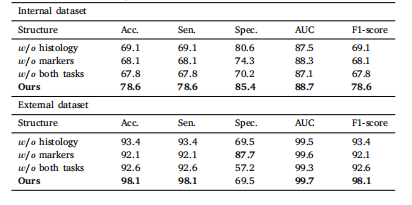
Table 7Mean values in terms of percentage for glioma classification accuracy by our methodwith and without auxiliary tasks.
表 7 本方法在有无辅助任务时的胶质瘤分类准确率(百分比平均值)


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









