





本文使用 Zhihu On VSCode 创作并发布
10.1 氦He (Helium)
10.1.1 氦的电离能
氦是比氢多一个电子的原子,但是计算更为复杂(没有解析解)。它的哈密顿量为:
这里取个近似。因为
在实验中我们可以得到氦的第一电离能和第二电离能,并据此可以计算出氦原子的基态能量。( 单位 1eV = 96.485 kJ/mol )
因此,我们可以得到氦原子的基态能量为-79eV。但是,根据类氢原子的公式
10.1.2 正氦和仲氦(Orthohelium & Parahelium)
在考虑氢的粗能级结构(gross structure)的时候并不需要考虑到电子的自旋。但是,对于氦原子,我们一开始就需要考虑到自旋的作用。当考虑到自旋作用之后,我们便可以把波函数分为空间部分和自旋部分。
根据泡利不相容原理,该波函数必须是交换反对称的,因此有两种可能:
具体写来为:
因此,我们把自旋为0的态叫做仲氦(parahelium),自旋为1的态作为正氦(orthohelium)。 在跃迁的时候,自旋不变。因为
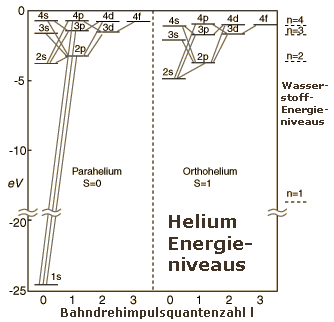
注意到,这里并没有
10.1.3 基态能量
我们希望在理论上得到与实验上相同的基态能量。这里有两个方法:微扰法和变分法。
实验测得微扰法变分法 -79eV-74.8eV-77.5eV可见,变分法具有比微扰法能准确的估计。这里分别进行介绍:
微扰法
微扰法非常简单,我们认为相互作用项为微扰,即
通过计算可以得到:
这里的
对于超出 r1 的部分,对势能没有贡献。之所以这样做是因为我们需要分解
因此,上述积分中只有k=0的球谐函数得以保留。用以方便计算,我们可以得到:
变分法
变分法是一种求基态能量的方法。利用一个带参数的试探波函数去得到
在这里,我们仍然采用1s1s态的波函数,参数为“核电荷数”Z。我们希望寻找更合适的Z。这里打引号是因为,我们并不改写哈密顿量,仅仅是让核电荷数为Z的波函数为试探波函数。
在这样的波函数下,去把哈密顿量表达成如下的形式更易计算:
根据类氢原子的公式,我们得到
对于第三项和微扰项一致,因此为
当,
进阶的试探波函数 Hylleraas method
Hylleraas 提出了新的坐标系。他给出
在这样的坐标下,他给出了如下形式的试探函数:
这里的k为有效电荷数,N为这里考虑的项数,项数越多约精准。他得到的能量为 -2.90324 a.u. 与实验数值 -2.90372 在小数点后3位都一致。
10.1.4 激发态能量
所谓的激发态,就是一个电子是1s态,另一个电子是nl态(仍是按照类氢原子的态划分)。这里的激发态只考虑了单电子激发,因为,双电子激发的能量要大于氢离子的能量。因此,会出现俄歇效应Auger effect:
而对于单电子激发态,能量不依赖于电子1或是2的标号,因此具有“交换兼并”(exchange degeneracy)的特性,所以,真实的波函数是这两个状态的线性组合。
仍然利用微扰法,我们得到,我们可以得到:
因此,通过解这个方程,我们得到了对称和反对称的空间波函数。和微扰能量。它由直接积分和交换积分组成。
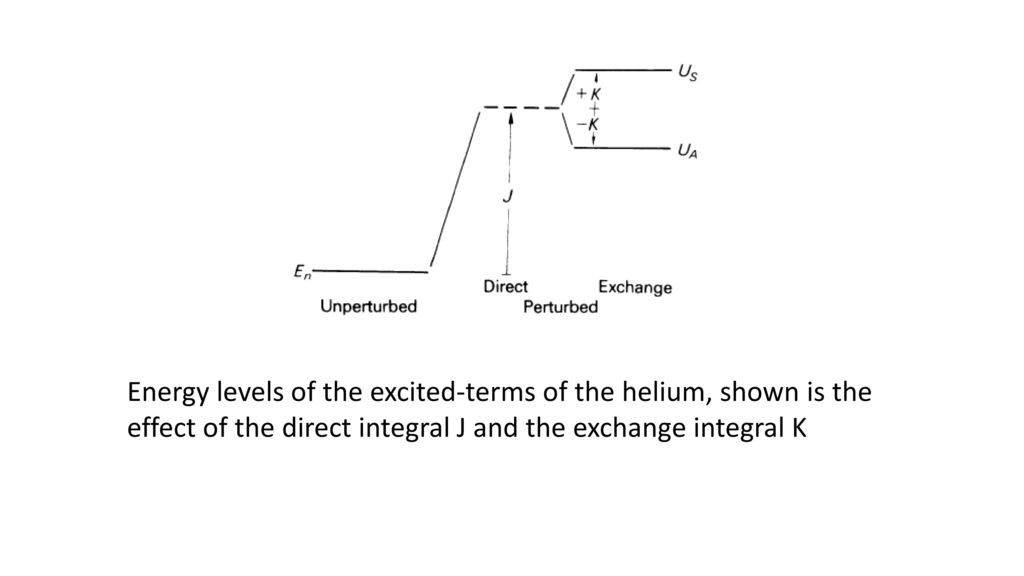
态随时间变化
接下来我们考虑态在时间作用下会如何变化。我们不妨让初态设为:
这里的正负态表达的是能量是J+K的态和J-K的态。因为他们的能量略有不同,因此,态的旋转的速率不同。
所以当
10.2 碱金属原子 (Alkali metal)
10.2.1 量子缺陷(Quantum Defacts)-- 碱金属光谱的概括
所谓碱金属,是指最外层1个电子,内部满壳层的情况。比惰性气体多出1个电子因此,它的电离能显著地低于惰性气体。对于碱金属的光谱来说,我们发现可以用如下的公式来表达就可以解释它的粗结构:
这里,
因此,量子缺陷常常总结成如下的式子:
下面,我们尝试去解释为什么会是这样(与n无关,与l有关,
10.2.2 有效核电荷数解释
我们是这样理解一个碱金属的:当最外层电子的离核距离比较远的时候,它感受到的核电荷数是1. 但是当它的离核距离比较进的时候(进入内层电子的区域)它会感受到更大的核电荷数直到Z。因此,它的势能是:
这里其实表达的是中心场近似(central-field approximation) .认为内部电子的分布是中心对称的,因此内层电子对价电子的作用也是中心对称的。于是,我们也会把这样的势能表达成:
因此在径向方向的分布函数是(类氢原子)
可以看到,整体的势能会带有一项“离心力项”(centrifugal barier), 它阻止了当
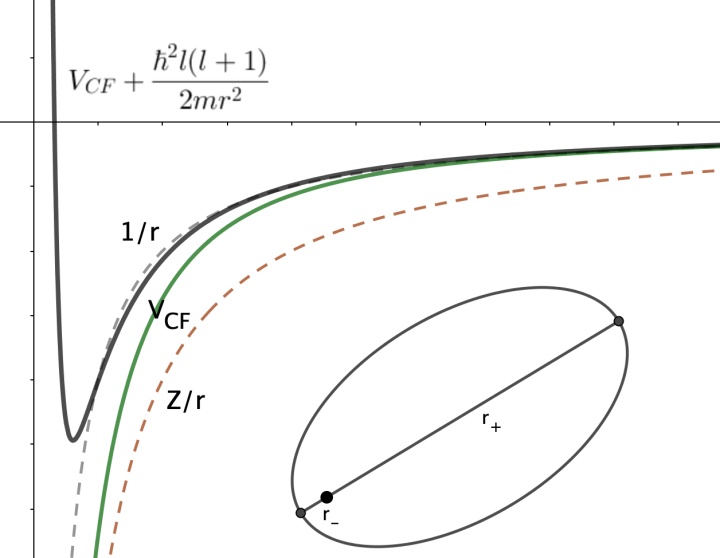
在这种近似下,我们发现,“近日点”只与l有关,“远日点”至于n有关。当l很小的时候,离核很近更受影响,而当l变大的时候离核越远。这里解释了为什么当l变大的时候量子缺席越来越小。
10.2.3 径向波函数解释
径向波函数渐进分析
我们仍然利用类氢原子波函数去理解碱金属原子的行为。首先定义一个碱金属原子可用的单位长度
因此,我们考虑

可以看到,s波具有3个波包,p波具有2个波包,d波具有1个波包。s波包在离核很近的时候具有更多的概率。同时,我们注意到,当 l 固定的时候,变化n,(
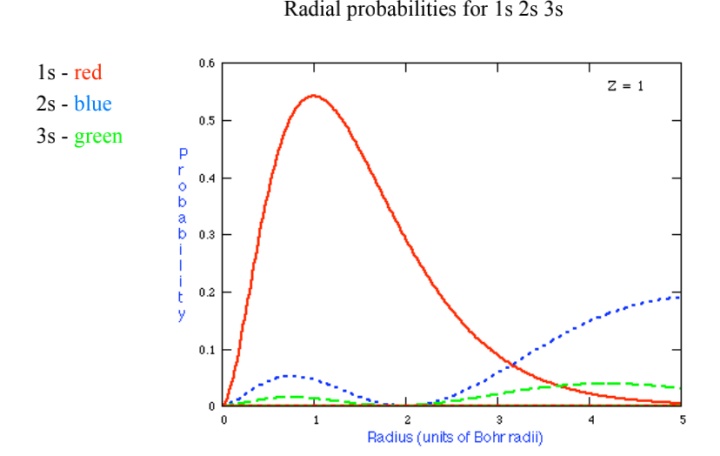
这也间接解释了为什么不同的n的量子缺陷是近乎相同的。
10.2.4 微扰论解释
因此,我们在类氢原子的哈密顿量基础上考虑微扰
不妨设,
10.3.4 相位解释
量子缺陷作为一个相位出现。仍然是利用类氢原子的方程去解释。径向方程为
我们这里取两个变量
在一般的氢原子解中,
我们希望
这时候我们希望把上式拓展到碱金属原子。在拓展到碱金属的时候就不仅仅有f形式的解了,也有一部分g函数形式。我们认为这两种形式的占比为
此时,带入















 4046
4046











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









