






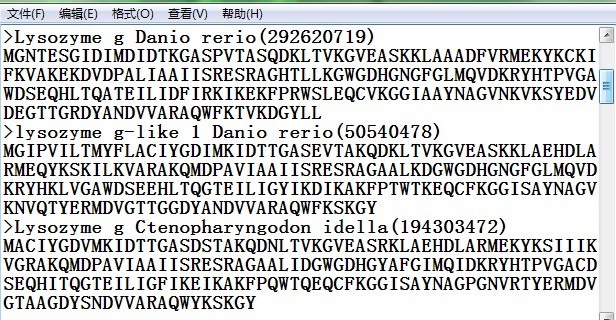
一、序列文本的准备
构树之前先将目标基因序列都分别保存为txt文本文件中(或者把所有序列保存在同一个txt文本中,可以用“>基因名称”作为第一行,然后重起一行 编辑基因序列),序列只包含序列字母(ATCG或氨基酸简写字母)。文件名名称可以已经您的想法随意编辑。

二、序列导入到Mega 5软件
(1)打开Mega 5软件,界面如下
(2)导入需要构建系统发育树的目的序列
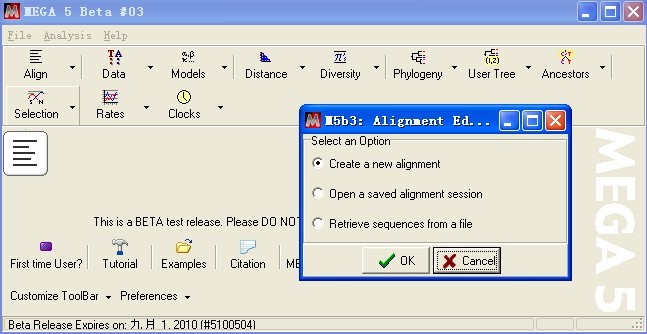
OK
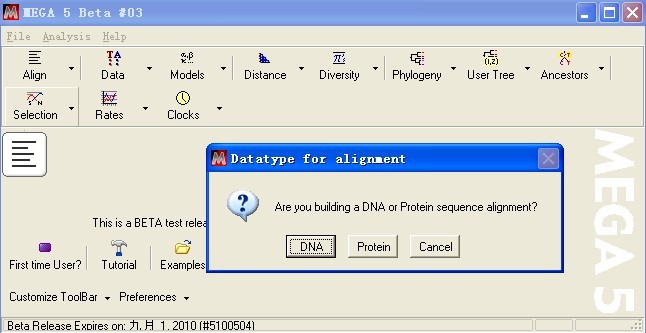
选择分析序列类型(如果是DNA序列,点击DNA,如果是蛋白序列,点击Protein)
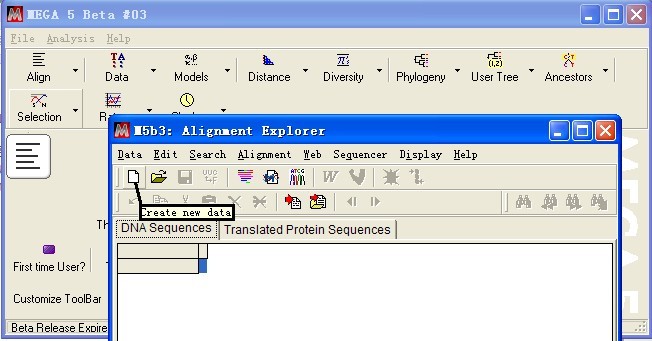
出现新的对话框,创建新的数据文件
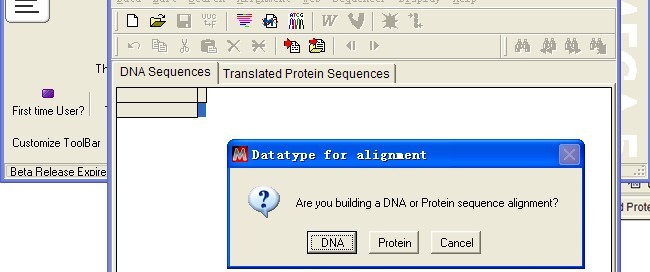
选择序列类型
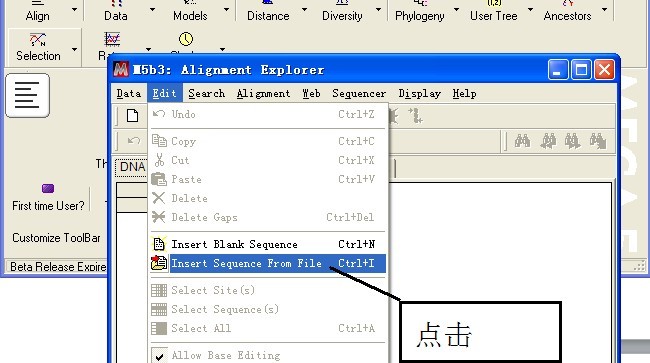
导入序列
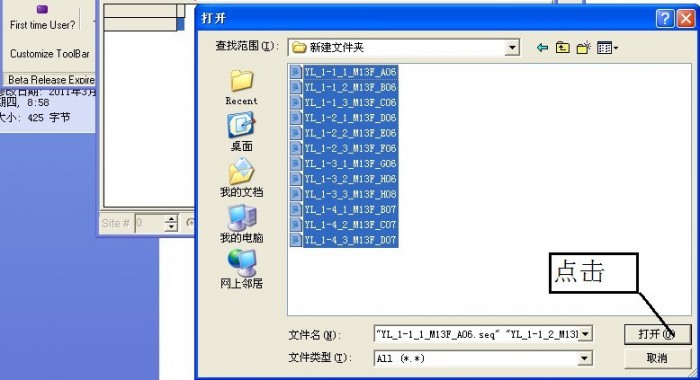
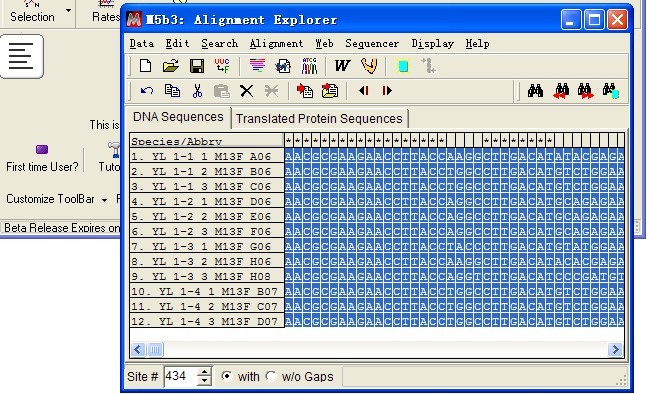
导入序列成功。
(3)序列比对分析
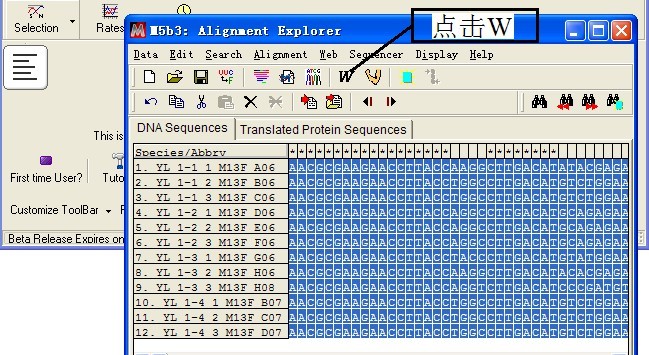
点击工具栏中“W”工具,进行比对分析,比对结束后删除两端不能够完全对齐碱基
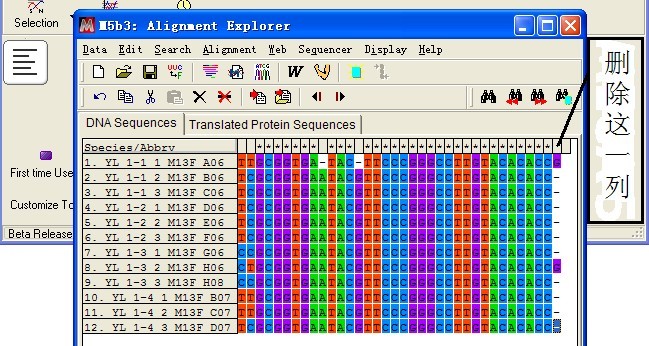
(4)系统发育分析
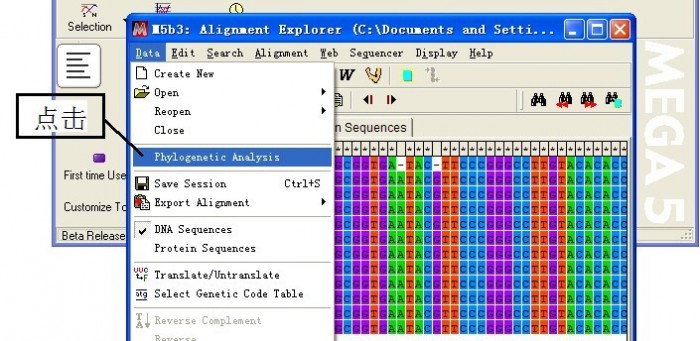
关闭窗口,选择保存文件路径,自定义文件名称
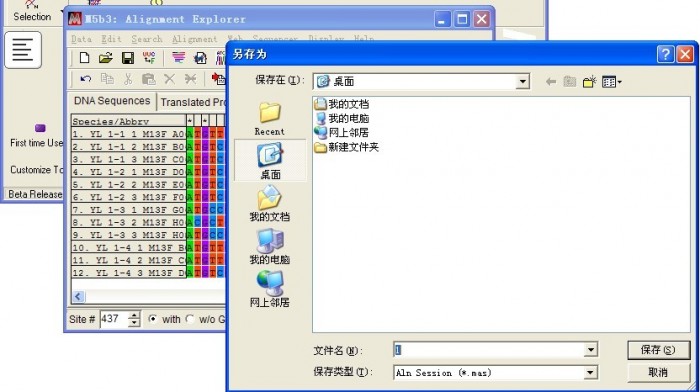
三、系统发育树构建
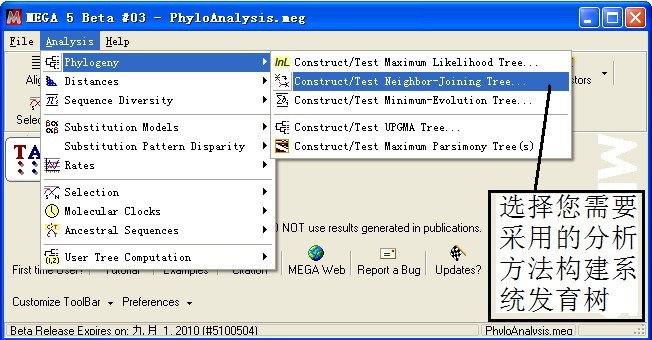
根据不同分析目的,选择相应的分析算法,本例子以N—J算法为例
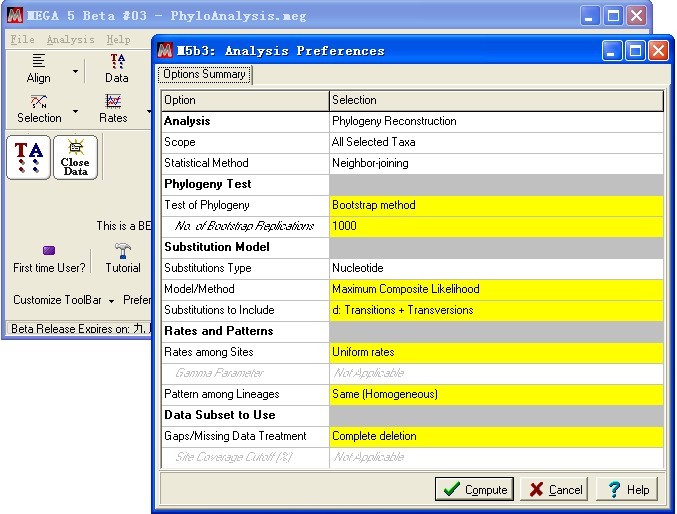
Bootstrap 选择1000,点击Compute,开始计算
计算完毕后,生成系统发育树。
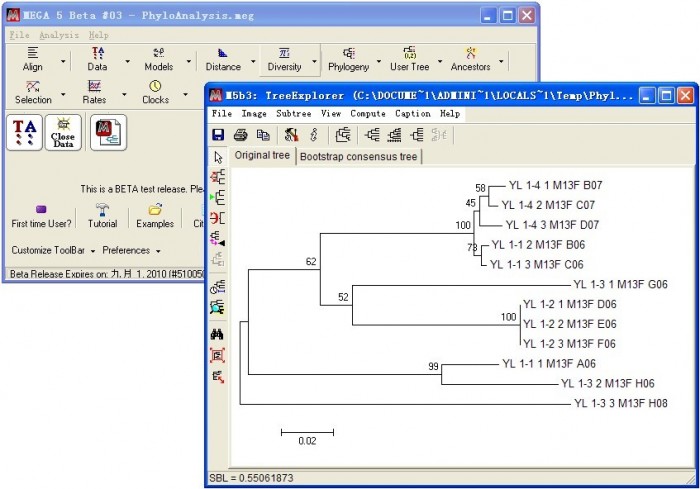
根据不同目的,导出分析结果,进行简单的修饰,保存
本方法来自网络,经小编microibs编辑,修改补充,如果转载请注明PLoB出处。















 1568
1568











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









