







 QuantumATK是一款强大的分子动力学模拟软件,支持DFT、ForceField等多种能量-力计算方法和动力学方法。其高效的动力学引擎和DFT+ForceField混合方法,适用于复杂材料系统的动力学模拟。特色功能包括time-stamped force-bias Monte Carlo、Metadynamics(PLUMED)和akMC方法,能进行长时间尺度和复杂过程的模拟。NanoLab图形界面使得研究者能轻松进行模拟和分析,提供丰富的计算和可视化工具。
QuantumATK是一款强大的分子动力学模拟软件,支持DFT、ForceField等多种能量-力计算方法和动力学方法。其高效的动力学引擎和DFT+ForceField混合方法,适用于复杂材料系统的动力学模拟。特色功能包括time-stamped force-bias Monte Carlo、Metadynamics(PLUMED)和akMC方法,能进行长时间尺度和复杂过程的模拟。NanoLab图形界面使得研究者能轻松进行模拟和分析,提供丰富的计算和可视化工具。
概述
动力学模拟是一种重要的原子级模拟方法,通过求解原子运动的经典力学牛顿方程对相空间进行采样,不仅可以研究体系在相空间的演化过程,还可以通过产生的系列结构(系综)通过统计方法得到体系在非零温度下的各种性质。 动力学过程中的原子间相互作用力则可以通过多种方法求得,可以是密度泛函理论,也可以是经验力场。
使用QuantumATK进行材料动力学模拟
- 可以用多种能量-力计算方法
- 密度泛函理论(DFT-LCAO 和 DFT-PlaneWave)
- 半经验量子力学模型(SemiEmpirical)
- 经验力场(Forcefield)
- 支持多种系综和理论方法
- NVE velocity verlet
- NPT with stress mask
- NPT/NVT(Berendsen)
- NPT Melchionna
- NPT with stress mask
- Langevin
- 支持多种动力学方法
- 平衡态分子动力学
- 非平衡态分子动力学(RNEMD)计算热导
- time-stamped force-bias Monte Carlo长时域的动力学方法
- Metadyamics(PLUMED):更快的对能量(自由能)面进行采样,获得大范围的结构-能量信息
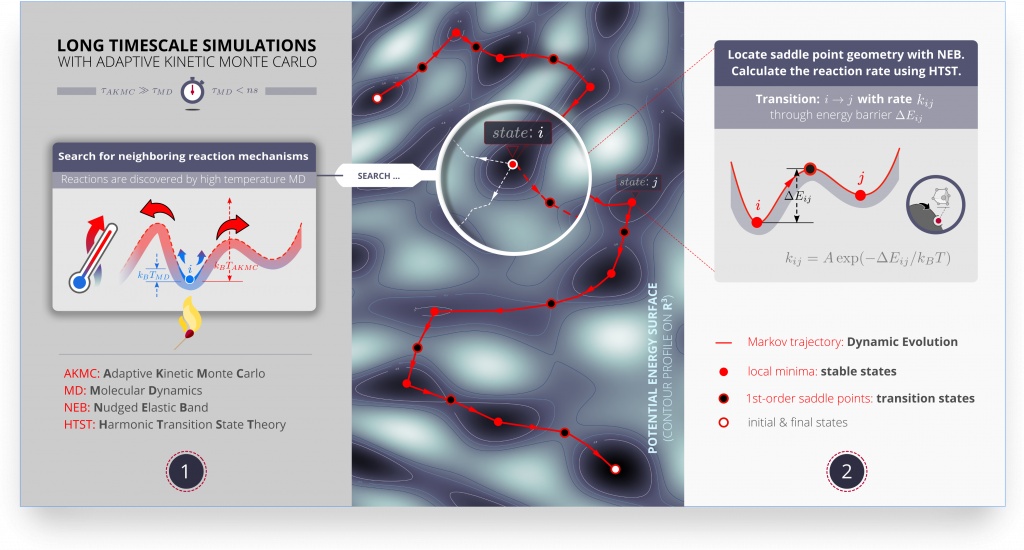
- 自适应动力学蒙特卡罗方法(adaptive kinetic Monte Carlo):研究结构变化与机理
- 可控制局域温度、设定升温速率
- 计算过程中分析
QuantumATK:高效的动力学引擎
基于QuantumATK高效的DFT、SE和ForceField计算引擎和MD代码的优化,分子动力学计算速度有明显优势。其中DFT、SE、ForceField均支持MPI大规模并行,并获得极大的速度提升。
基于DFT的MD计算速度测试结果7056原子的分子动力学(水分子):在64MPI并行时,第一步MD耗时4分钟。

3220原子的分子动力学(固
 最低0.47元/天 解锁文章
最低0.47元/天 解锁文章


















 585
585

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









