







一、概念辨析
(1)一致性
一致性(identity):如果两个序列(蛋白质或核酸)长度相同,那么它们的一致性定义为它们对应位置上相同的残基(一个字母,氨基酸或碱基)的数目占总长度的百分数,即
identity =(一致字符的个数/全局比对长度)*100%
(2)相似性
相似性(similarity):如果两个序列(蛋白质或核酸)长度相同,那么它们的相似度定义为它们对应位置上相似的残基与相同的残基的数目和占总长度的百分数,残基两两相似的量化关系被替换记分矩阵所定义,即
similarity=(一致及相似的字符个数/全局比对长度)*100%
做法:先做双序列全局/局部比对,再根据比对结果和比对长度计算一致性和相似性
言简意赅:
identity指的是对应位置上相同残基的数目占总长度的百分数。
similarity指的是对应位置上相同和相似的参基占总数的百分数。
一般用相似性和一致性推测两个序列的同源性 (好像identity>30%,similarity>50%可以说是同源的,没有明确的量化指标,根据自己研究的问题决定阈值大小)。
同源性(homology)是一个进化学上的概念,没有程度,只有同源或者不同源。所以说两个序列95%同源,或者高度同源都是不对的。
二、替换记分矩阵
替换记分矩阵是一个生物信息学的概念。替换记分矩阵是反映残基之间相互替换率的矩阵。也就是说,它描述了残基两两相似的量化关系。DNA序列有DNA序列的替换记分矩阵,蛋白质序列有蛋白质序列的替换记分矩阵,不可混淆使用。
DNA序列的替换记分矩阵主要有三种,分别是等价矩阵,转换-颠换矩阵,BLAST矩阵。
蛋白质序列的替换记分矩阵主要等价矩阵,PAM矩阵,BLOSUM矩阵等。其中BLOSUM62是应用得最广的氨基酸替换矩阵,是BLAST中蛋白质序列比对的默认矩阵。
BLOSUM矩阵的推导过程见知乎:
三、计算Identity和Similarity
这里我们隆重推出一个可视化工具:EMBOSS Water < EMBL-EBI,EMBOSS Water使用Smith-Waterman算法来计算两个序列的局部对齐,全局对齐使用的是 EMBOSS Needle < EMBL-EBI。
Smith-Waterman算法的具体推导过程参考Wikipedia:Smith–Waterman algorithm - Wikipedia

EMBOSS Water除了在线版还有本地版。我们有很多对的蛋白序列需要计算相似性,因此在本次实验中我们使用本地版批量运行。我尝试了在Windows系统上安装,不成功,所以推荐大家在Lnux环境中用Conda安装,这样会少走一些弯路。参考:Emboss | Anaconda.org
conda install bioconda::emboss
或者
conda install bioconda/label/cf201901::emboss安装成功之后,water -h 查看一下是否成功安装上。
(MyEnv) qfchen@Tanlab-Server:~$ water -h
Smith-Waterman local alignment of sequences
Version: EMBOSS:6.6.0.0
Standard (Mandatory) qualifiers:
[-asequence] sequence Sequence filename and optional format, or
reference (input USA)
[-bsequence] seqall Sequence(s) filename and optional format, or
reference (input USA)
-gapopen float [10.0 for any sequence] The gap open penalty
is the score taken away when a gap is
created. The best value depends on the
choice of comparison matrix. The default
value assumes you are using the EBLOSUM62
matrix for protein sequences, and the
EDNAFULL matrix for nucleotide sequences.
(Number from 0.000 to 100.000)
-gapextend float [0.5 for any sequence] The gap extension
penalty is added to the standard gap penalty
for each base or residue in the gap. This
is how long gaps are penalized. Usually you
will expect a few long gaps rather than many
short gaps, so the gap extension penalty
should be lower than the gap penalty. An
exception is where one or both sequences are
single reads with possible sequencing
errors in which case you would expect many
single base gaps. You can get this result by
setting the gap open penalty to zero (or
very low) and using the gap extension
penalty to control gap scoring. (Number from
0.000 to 10.000)
[-outfile] align [*.water] Output alignment file name
(default -aformat srspair)
Additional (Optional) qualifiers:
-datafile matrixf [EBLOSUM62 for protein, EDNAFULL for DNA]
This is the scoring matrix file used when
comparing sequences. By default it is the
file 'EBLOSUM62' (for proteins) or the file
'EDNAFULL' (for nucleic sequences). These
files are found in the 'data' directory of
the EMBOSS installation.
Advanced (Unprompted) qualifiers:
-[no]brief boolean [Y] Brief identity and similarity
General qualifiers:
-help boolean Report command line options and exit. More
information on associated and general
qualifiers can be found with -help -verbose接下来是批量使用的water的代码,事先要将序列保存成单个fasta文件:
import os
import subprocess
input_folder = '/home/qfchen/Water/fasta/'
output_folder = '/home/qfchen/Water/results/'
os.makedirs(output_folder, exist_ok=True)
fasta_files = [f.split('.')[0] for f in os.listdir(input_folder)]
for file1 in fasta_files:
for file2 in fasta_files:
if file1 != file2:
file_a = os.path.join(input_folder, file1 + '.fasta')
file_b = os.path.join(input_folder, file2 + '.fasta')
output_file = os.path.join(output_folder, f"{file1}_{file2}.water")
cmd = [
"water",
"-auto",
"-asequence", file_a,
"-bsequence", file_b,
"-datafile", 'EBLOSUM62',
"-gapopen", "10.0",
"-gapextend", "0.5",
"-outfile", output_file]
print(f"Running: {' '.join(cmd)}")
try:
subprocess.run(cmd, check=True)
print(f"Alignment complete: {output_file}")
except subprocess.CalledProcessError as e:
print(f"Error running water for {file_a} and {file_b}: {e}")运行完毕的结果是txt文件,下面是展示的结果,我们的目的是解析这个txt文件,获取里面的identity和similarity。
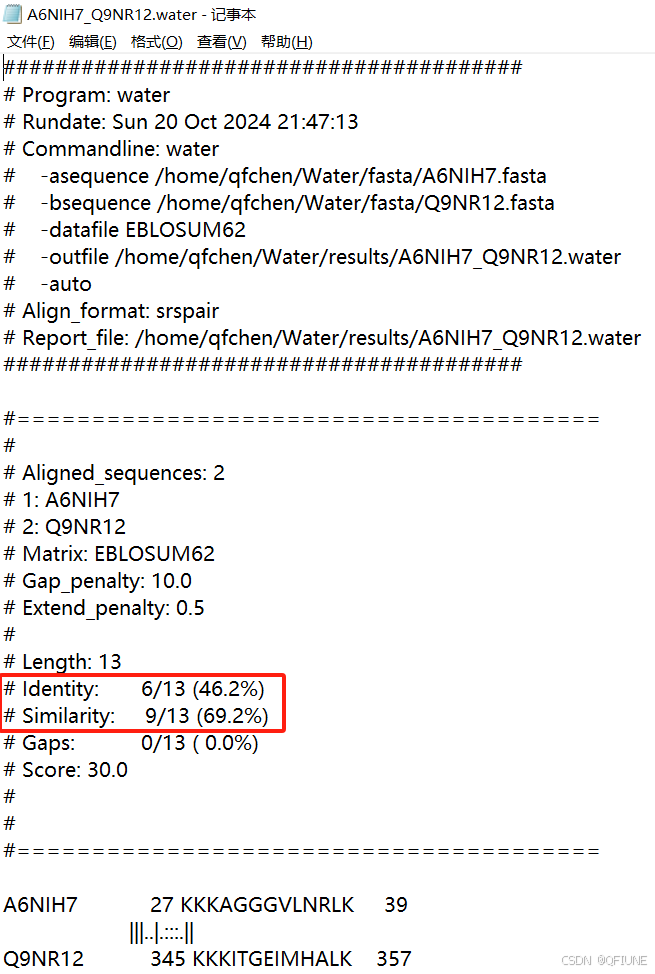
解析代码如下:
import re
import os
import pandas as pd
# 定义要解析的文本文件路径
input_dir = '/home/qfchen/Water/results'
for root, dirs, files in os.walk(input_dir):
all_results = []
for file in files:
pair = file.split('.')[0]
prot1 = pair.split('_')[0]
prot2 = pair.split('_')[1]
input_file = os.path.join(root, file)
identity = None
similarity = None
# 读取文件并提取 Identity 和 Similarity 值
with open(input_file, 'r') as file:
for line in file:
# 使用正则表达式查找Identity行
if line.startswith("# Identity:"):
identity_match = re.search(r"(\d+\.?\d*)%", line)
if identity_match:
identity = identity_match.group(1) # 提取数字部分
# 使用正则表达式查找Similarity行
if line.startswith("# Similarity:"):
similarity_match = re.search(r"(\d+\.?\d*)%", line)
if similarity_match:
similarity = similarity_match.group(1) # 提取数字部分
all_results.append([pair, prot1, prot2, identity, similarity])
if identity and similarity:
print(f"Identity: {identity}%")
print(f"Similarity: {similarity}%")
else:
print("Identity or Similarity information not found!")
df = pd.DataFrame(all_results, columns=['Pairs', 'Protein1', 'Protein2', 'Identity', 'Similarity'])
df.to_csv('/home/qfchen/Water/all_results.csv')下面展示的部分解析结果:

四、一个坑
刚开始的时候,我在网上搜到了一个python包:swalign,参考了这个教程Python 中的 Smith-Waterman 算法 | D栈 - Delft Stack 用pip安装。
pip3 install swalign源码报错,于是乎就去Github下载源码手动安装:
pip3 install swalign
Collecting swalign
Using cached swalign-0.3.7.tar.gz (7.8 kB)
Preparing metadata (setup.py) ... error
error: subprocess-exited-with-error
× python setup.py egg_info did not run successfully.
│ exit code: 1
╰─> [6 lines of output]
Traceback (most recent call last):
File "<string>", line 2, in <module>
File "<pip-setuptools-caller>", line 34, in <module>
File "C:\Users\17608\AppData\Local\Temp\pip-install-z3fmqvft\swalign_c447e6fdada946b99600d5aff81a6e72\setup.py", line 4, in <module>
long_description = fh.read()
UnicodeDecodeError: 'gbk' codec can't decode byte 0xa6 in position 537: illegal multibyte sequence
[end of output]
note: This error originates from a subprocess, and is likely not a problem with pip.
error: metadata-generation-failed
× Encountered error while generating package metadata.
╰─> See above for output.
note: This is an issue with the package mentioned above, not pip.
hint: See above for details.git clone https://github.com/mbreese/swalign.git安装的时候先去把setup.py文件里的 with open("README.md", "r") as fh
改成 with open("README.md", "r", encoding='utf-8') as fh
然后在命令行里运行:
python setup.py install这样就成功安装了,下面是使用代码:
import swalign
# 定义匹配和不匹配的分数
match_score = 2
mismatch_score = -1
# 创建一个核苷酸评分矩阵
scoring = swalign.NucleotideScoringMatrix(match_score, mismatch_score)
# 创建一个局部对齐对象
alignment = swalign.LocalAlignment(scoring)
# 定义要比较的两个序列
sequence1 = "MTMDKSELVQKAKLAEQAERYDDMAAAMKAVTEQGHELSNEERNLLSVAYKNVVGARRSSWRVISSIEQKTERNEKKQQMGKEYREKIEAELQDICNDVLELLDKYLIPNATQPESKVFYLKMKGDYFRYLSEVASGDNKQTTVSNSQQAYQEAFEISKKEMQPTHPIRLGLALNFSVFYYEILNSPEKACSLAKTAFDEAIAELDTLNEESYKDSTLIMQLLRDNLTLWTSENQGDEGDAGEGEN"
sequence2 = "MDDREDLVYQAKLAEQAERYDEMVESMKKVAGMDVELTVEERNLLSVAYKNVIGARRASWRIISSIEQKEENKGGEDKLKMIREYRQMVETELKLICCDILDVLDKHLIPAANTGESKVFYYKMKGDYHRYLAEFATGNDRKEAAENSLVAYKAASDIAMTELPPTHPIRLGLALNFSVFYYEILNSPDRACRLAKAAFDDAIAELDTLSEESYKDSTLIMQLLRDNLTLWTSDMQGDGEEQNKEALQDVEDENQ"
# 执行对齐
results = alignment.align(sequence1, sequence2)
# 答应全部输出结果
print(results.dump())
# 获取一致性的分数
identity = results.identity
print(identity)这个Python包确实很方便,但是它里面没有相似性的计算功能,因此最后没有被采用。
参考资料
- https://blog.csdn.net/HuangXinyue1017/article/details/112437225
- https://www.jianshu.com/p/7b9a7c8c0a2d
- BLOSUM矩阵的推导 - 知乎 (zhihu.com)
- Smith-Waterman-Algorithm | WjWei | Blog

















 2176
2176

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









