






利用BWA软件进行比对之后,可以利用Samtools进行比对率查看,是检测分析结果的一个常用方法。
0.1 将测序时候两个分开的序列合并,有时候一次不能测够数据量,所以将两次的数据合并一下
cat B5_L1.R1.fastq.gz B5_L4.R1.fastq.gz > B5_L0_R1.fastq.gz代码:
Samtools flagstat -@ 8 392F.bam # @代表线程数结果:
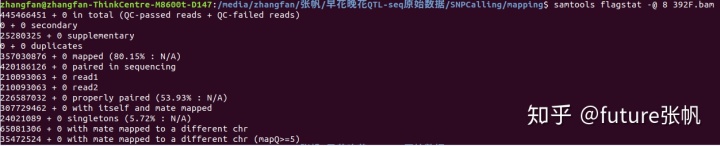
结果解释:
445466451 + 0 in total (QC-passed reads + QC-failed reads)
#质控通过的reads数+质控未通过的reads数,0x200 bit set 后面都是用这个参数区分的
0 + 0 secondary
#次级read数,0x100 bit set (不是很懂怎么算的)
25280325 + 0 supplementary
#额外reads数, 0x800 bit set
0 + 0 duplicates
#重复reads数,0x400 bit set
357030876 + 0 mapped (80.15% : N/A)
#匹配率,这个值比较重要,0x4 bit not set
420186126 + 0 paired in sequencing
#成对的序列数量,0x1 bit set
210093063 + 0 read1
#read1的数量,both 0x1 and 0x40 bits set
210093063 + 0 read2
#read2的数量,both 0x1 and 0x80 bits set
226587032 + 0 properly paired (53.93% : N/A)
#唯一匹配的reads数量,both 0x1 and 0x2 bits set and 0x4 bit not set
307729462 + 0 with itself and mate mapped
#非唯一匹配的reads数量,0x1 bit set and neither 0x4 nor 0x8 bits set
24021089 + 0 singletons (5.72% : N/A)
#只有一条染色体匹配上参考基因组的数量,both 0x1 and 0x8 bits set and bit 0x4 not set
65081306 + 0 with mate mapped to a different chr
#比对到不同染色体的reads数,0x1 bit set and neither 0x4 nor 0x8 bits set and MRNM not equal to RNAME
35472524 + 0 with mate mapped to a different chr (mapQ>=5)
#比对到不同染色体,但是比对质量值大于5的reads数,0x1 bit set and neither 0x4 nor 0x8 bits set and MRNM not equal to RNAME and MAPQ >= 5
总结:这个参数还是比较多的,最主要的是比对率和唯一比对率,其他的参数都是依据bits set区分的(不是很懂),主要是通过bam文件中的前13个序列信息字符来确定的。
参考资料:
https://blog.csdn.net/u013553061/article/details/53402232blog.csdn.net http://www.htslib.org/doc/samtools.htmlwww.htslib.org














 1959
1959











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









