









项目结构:
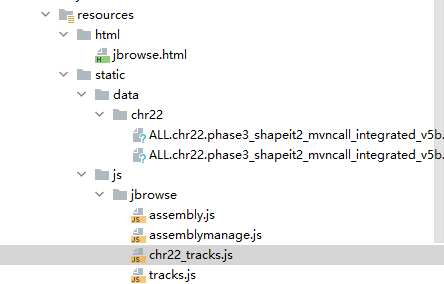
html:
<h1>We're using JBrowse Linear View!</h1>
<div>
<div id="jbrowse_linear_view" style="min-height:700px;"></div>
<script type="module" src="../static/js/jbrowse/assemblymanage.js" ></script>
</div>
assemblymanage.js:
import * as genome from './assembly.js'
var variant_tracks = new Array()
//chr22
import * as chr22_tracks from './chr22_tracks.js'
variant_tracks['chr22'] = chr22_tracks
variant_tracks['chr22_seq']=genome.chr22
//初始界面设定
var tracks = variant_tracks['chr22'].tracks
var assembly = variant_tracks['chr22_seq']
var genomeView = new JBrowseLinearGenomeView({
container: document.getElementById('jbrowse_linear_view'),
assembly,
tracks
// location: '22:10,210..21,798',
})
assembly.js:(uri地址错误,可下载放在项目里)
export var chr22 = {
name:"chr22",
sequence:{
type:'ReferenceSequenceTrack',
trackId:'chr22_ReferenceSequenceTrack',
adapter:{
type: 'BgzipFastaAdapter',
fastaLocation: {
uri: "http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/technical/reference/phase2_reference_assembly_sequence/hs37d5.fa.gz"
},
faiLocation: {
uri: "http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/technical/reference/phase2_reference_assembly_sequence/hs37d5.fa.gz.fai"
},
gziLocation: {
uri: "http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/technical/reference/phase2_reference_assembly_sequence/hs37d5.fa.gz.gzi"
}
}
}
}
chr22_tracks.js:(目前只展示chr22)
export var tracks = [
{
type:'VariantTrack',
trackId:'ALL.chr22.phase3_shapeit2_mvncall_integrated_v5b.20130502.genotypes.vcf',
name:"1000 Genomes Variant Calls CHR",
category:['1000Genomes','Variants'],
assemblyNames:['chr22'],
adapter:{
type: 'VcfTabixAdapter',
vcfGzLocation:{
uri:
'../static/data/chr22/ALL.chr22.phase3_shapeit2_mvncall_integrated_v5b.20130502.genotypes.vcf.gz',
},
index:{
location:{
uri:
'../static/data/chr22/ALL.chr22.phase3_shapeit2_mvncall_integrated_v5b.20130502.genotypes.vcf.gz.tbi',
},
indexType:'TBI',
}
}
}
]
页面效果图:
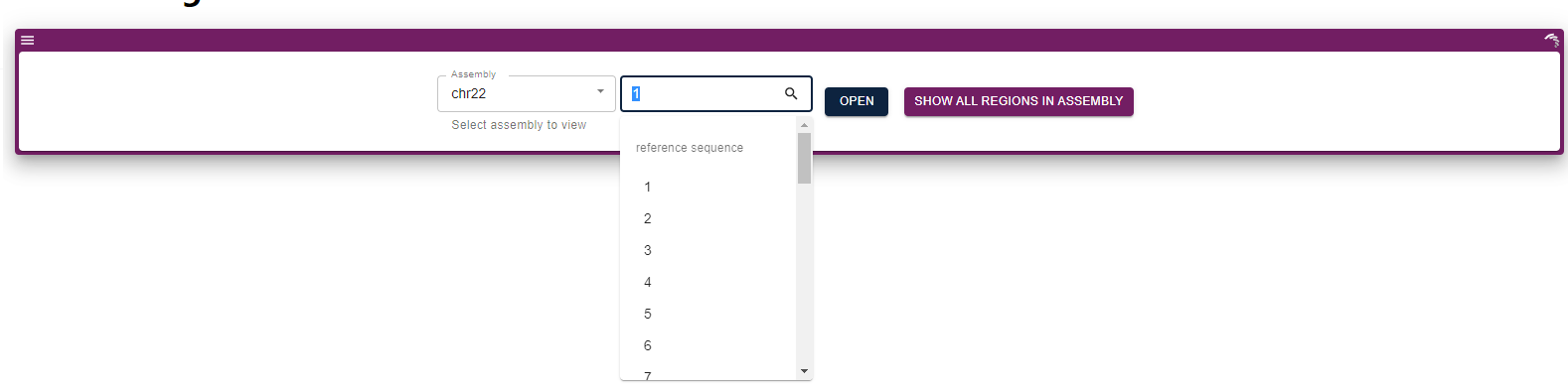
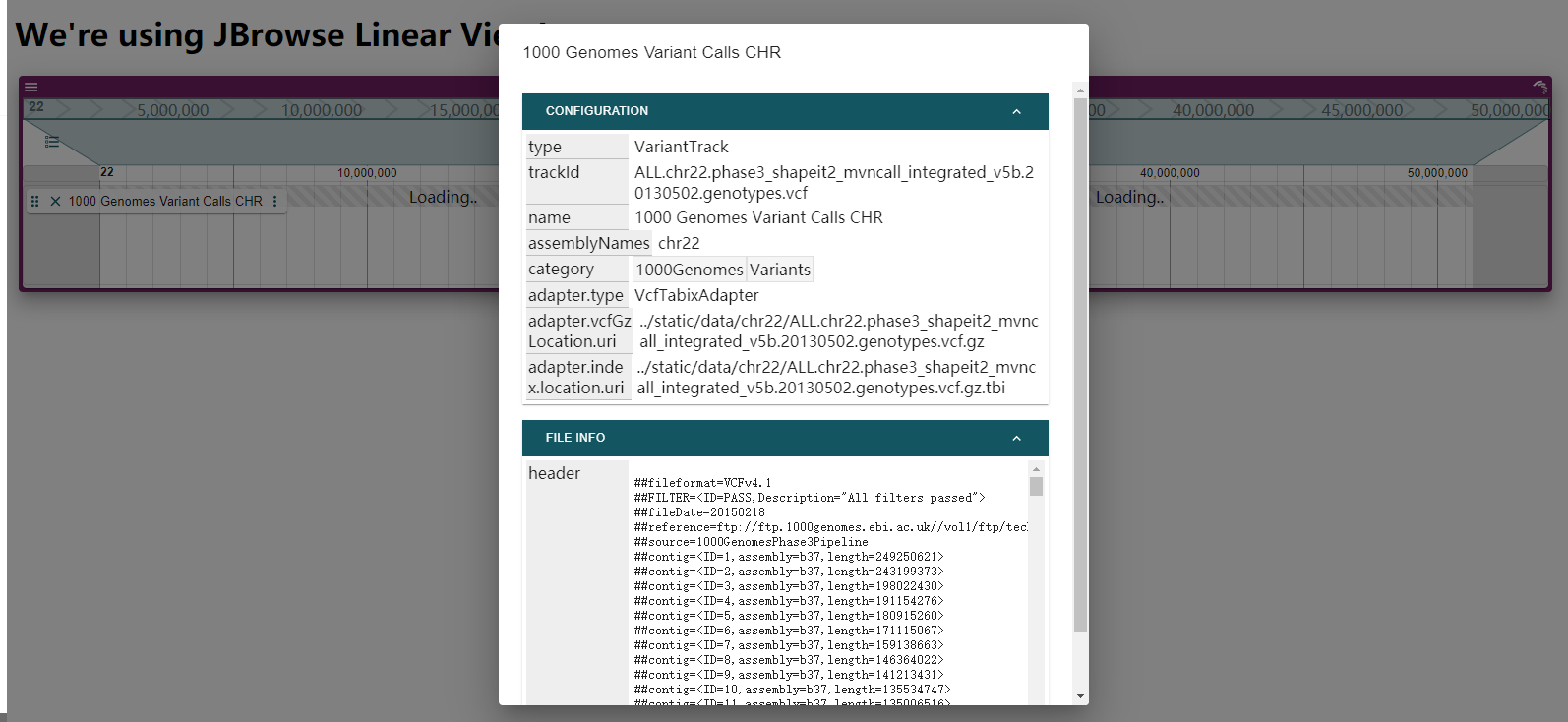
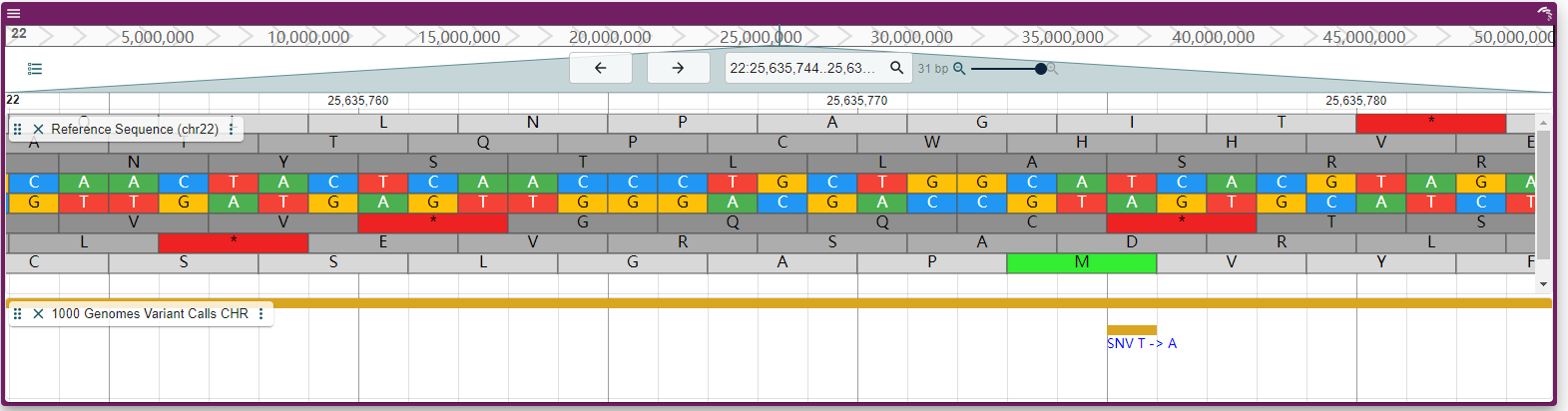
待续…















 6109
6109











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









