







shell conda命令安装:
conda install -c bioconda vcftools
vcftools文档:
OUTPUT FILE OPTIONS
--out <output_prefix>
This option defines the output filename prefix for all files generated by vcftools. For example, if <prefix> is set to output_filename, then all output files will be of the form output_filename.*** . If this option is omitted, all output files will have the prefix "out." in the current working directory.
--stdout
-c
These options direct the vcftools output to standard out so it can be piped into another program or written directly to a filename of choice. However, a select few output functions cannot be written to standard out.
--temp <temporary_directory>
This option can be used to redirect any temporary files that vcftools creates into a specified directory.
ALLELE FILTERING
--maf <float>
--max-maf <float>
Include only sites with a Minor Allele Frequency greater than or equal to the "--maf" value and less than or equal to the "--max-maf" value. One of these options may be used without the other. Allele frequency is defined as the number of times an allele appears over all individuals at that site, divided by the total number of non-missing alleles at that site.
OUTPUT VCF FORMAT
--recode
--recode-bcf
These options are used to generate a new file in either VCF or BCF from the input VCF or BCF file after applying the filtering options specified by the user. The output file has the suffix ".recode.vcf" or ".recode.bcf". By default, the INFO fields are removed from the output file, as the INFO values may be invalidated by the recoding (e.g. the total depth may need to be recalculated if individuals are removed). This behavior may be overriden by the following options. By default, BCF files are written out as BGZF compressed files.
--recode-INFO <string>
--recode-INFO-all
These options can be used with the above recode options to define an INFO key name to keep in the output file. This option can be used multiple times to keep more of the INFO fields. The second option is used to keep all INFO values in the original file.
根据ALLELE FREQUENCY过滤,0.001 < AF < 0.5,保留INFO域信息:
$ vcftools --vcf ALL.chr22.phase3_shapeit2_mvncall_integrated_v5b.20130502.genotypes.vcf --maf 0.001 --max-maf 0.5 --recode --recode-INFO-all --out chr22_af_filter
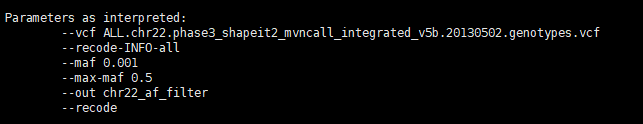
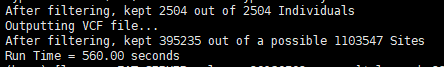
输出文件:
注意:
–maf,–max-maf Minor Allele Frequency二等位基因频率进行过滤,常为–maf 0.05,保留大于0.05的。
–non-ref-af,–non-ref-ac… 保留都是ALT变异的位点。
–mac INT,–max-mac 保留Minor Allel
Count数大于INT数的位点
–min-alleles 2, --max-alleles 2筛选保留含有2个ALT变异的位点。常用。















 621
621











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









