







 本文介绍了MaterialsStudio软件的基础入门,包括分子模拟概述、软件特点、版本选择与安装、显示器设置、快捷键使用以及五个实际案例,如有机分子吸附、缓蚀剂研究、MOF吸附等,帮助读者掌握MS软件的使用。
本文介绍了MaterialsStudio软件的基础入门,包括分子模拟概述、软件特点、版本选择与安装、显示器设置、快捷键使用以及五个实际案例,如有机分子吸附、缓蚀剂研究、MOF吸附等,帮助读者掌握MS软件的使用。
【Materials Studio】入门基础篇
前言
本文是对学习MS软件时,对视频的学习总结,无任何商务行为。视频链接如下:
https://www.bilibili.com/video/BV1SZ4y1o73r/?spm_id_from=333.999.0.0

知识准备:分子模拟简介

经典的分子模拟过程
- 构建模型;
- 设置计算参数;
- 运行计算;
- 分析与展示计算结果。
完成上述4个步骤,即完成了一次分子模拟。
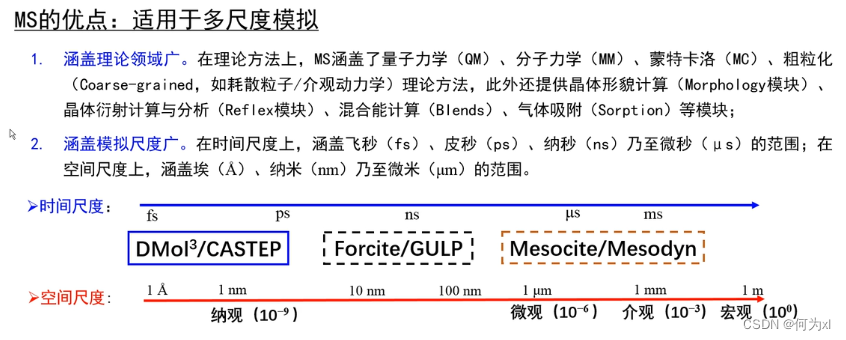
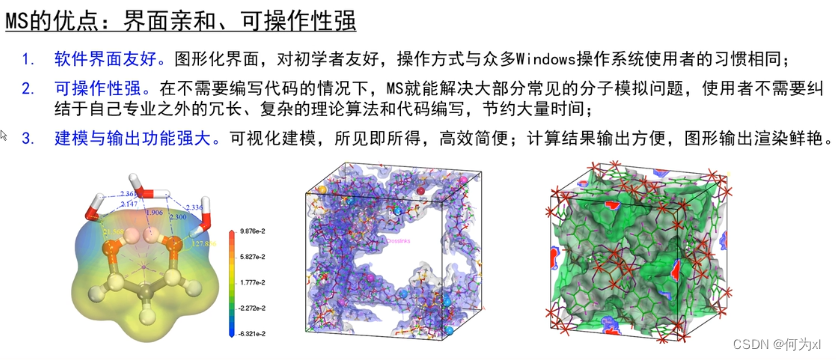
MS软件的特点和优势


MS模块选择的经验原则






https://www.3ds.com/products-services/biovia/references/
三键鼠标
二、MS 软件的版本选择、安装、存放路径
MS 软件的版本选择、安装、存放路径

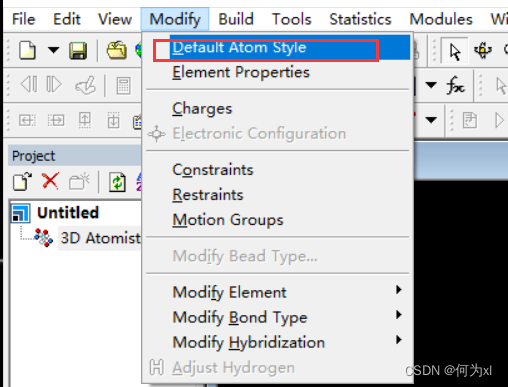
MS软件使用的基本设置

显示器设置
初始界面:
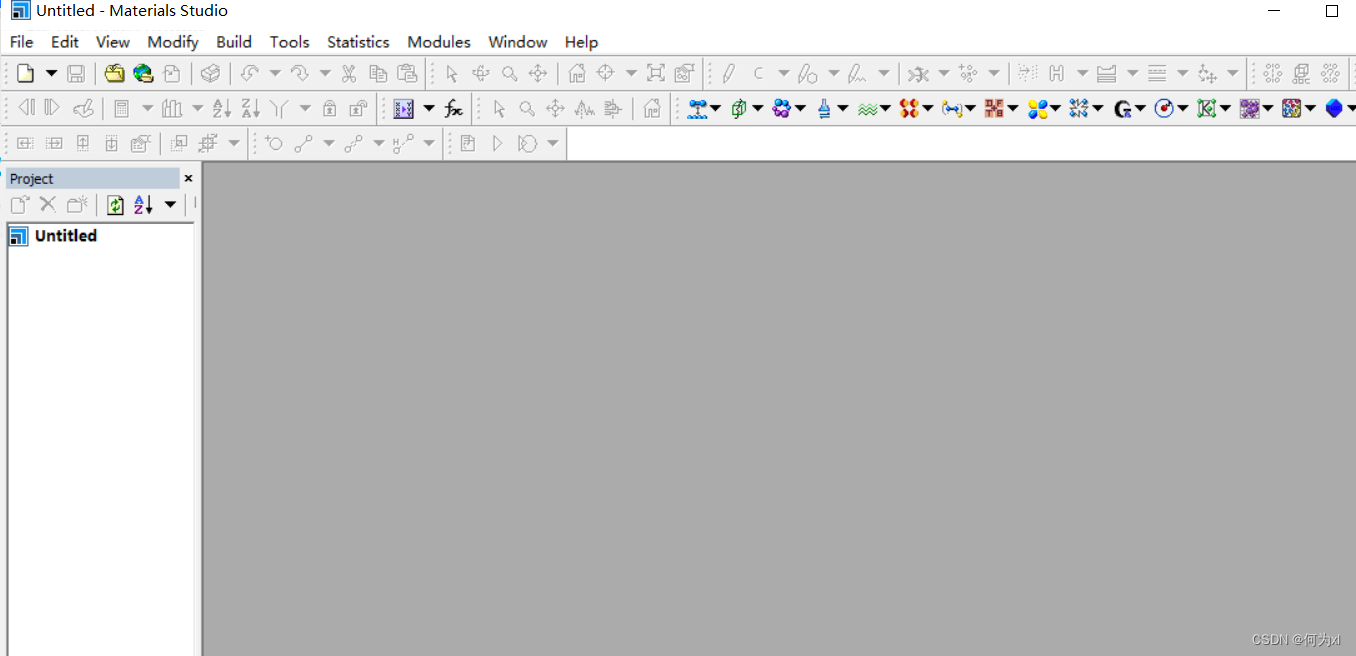
点击 View -> Explorers ->Job Explorer (Properties Explorer) 注意这两个都要点
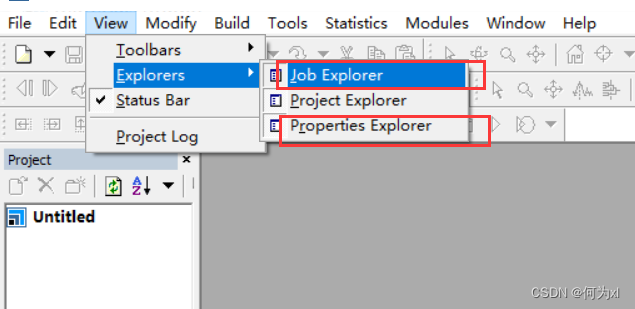
推荐修改的显示器界面(从图中可以看到 MS 界面里面需要有三个管理器)
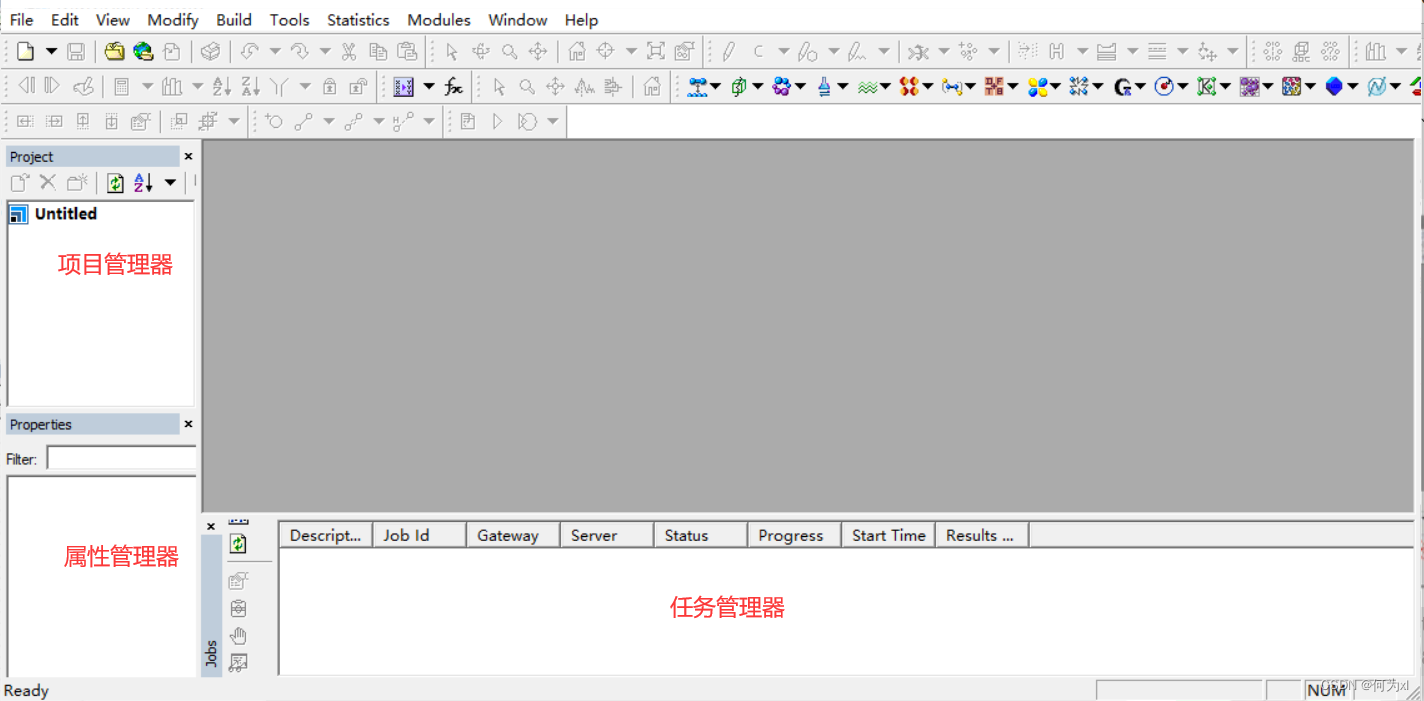
还原MS默认的参数设置((每次新建project*时
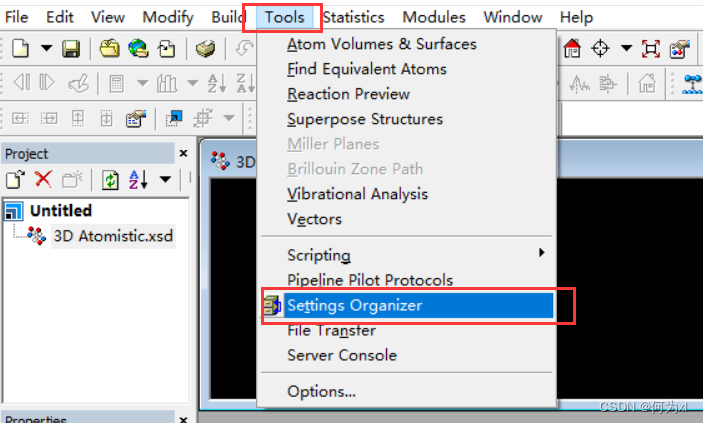
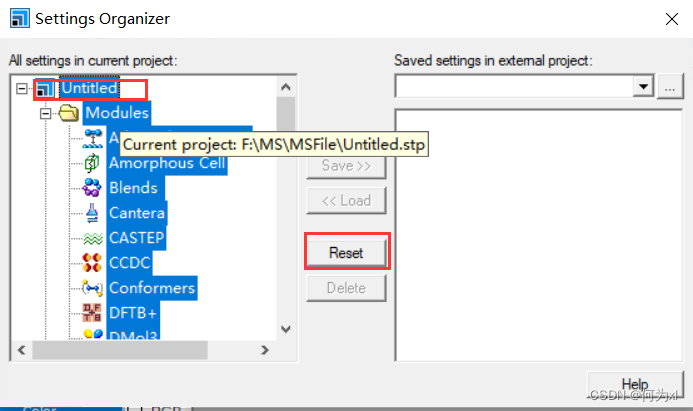
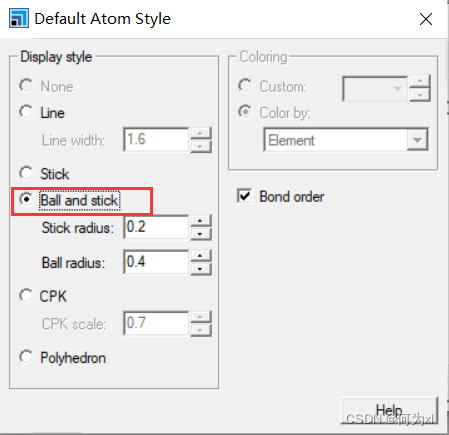
模型默认显示模式设置(每次新建project时))
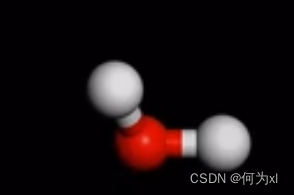
默认的显示模式(line 线性),下图如默认形式下的水分子
MS 快捷键的使用
通过鼠标框设置:
ctrl + A:会把键也同样选中
alt + 鼠标点击 = 选择所有相同的原子
shift + 中键 = 局部模型旋转
home + 箭头 = 各类视图
通过算例认识 MS
建模十分重要。基础中的基础。

案例一、有机分子在金属氧化物表面的吸附与分解
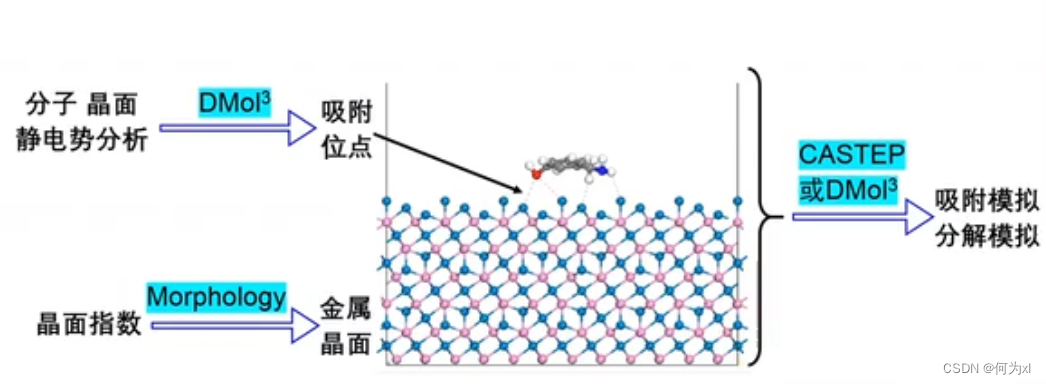
做模拟:
第一步建模;金属氧化物表面,有“表面”二字,就意味着我们需要来做切面,即做晶面切割,这里需要知道晶面指数。旧的晶体可以通过文献进行查看,但是涉及到新的晶体,如何确定新晶体所具有的代表性的晶面指数。(这是一个非常常见与非常普遍的问题)
解决方法:使用 MS 的模块 —— Morphology
这个模块可以算出来任意晶体的代表性的晶面指数
将晶面指数输入后,可以获得晶体模型,获得晶面模型之后下一步是要把分子给它放上去,(新问题:分子该怎么放,放在什么位置?)
我们可以徳用模抉DMol3来分别计算分子和晶体的静电势。
然局根据电荷同性相后、异性相吸的原理来摆放一个良好的初始位置。
模型有了之后,我们下一步就可以使用CASTEP 或者DMol3 来做模拟了
通过三个模块之间的协作,从而使得这个模拟更加容易和顺利。
案例二、缓蚀剂对金属表面缓蚀机理的研究

案例三、 MOF 吸附气体分子的研究
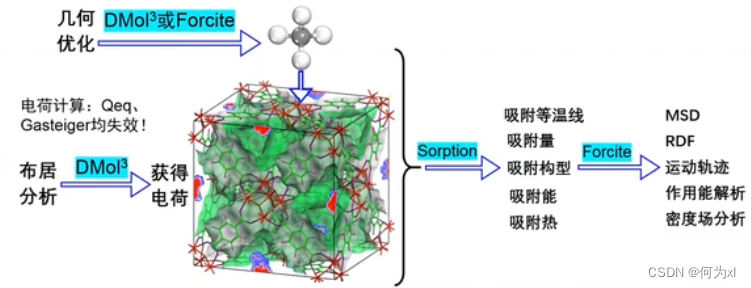
基于分子力学,即使用力场来进行计算。
进行吸附之前,将力场和电荷设置好。但是对于 MOF 如果使用分子力学常用的,诸如 Qeq 这种算法来算,会失效。
使用 Sorption 来做吸附计算,可以算出来 吸附量、吸附等温线、吸附能。
案例四、环氧固化产物的热解模拟
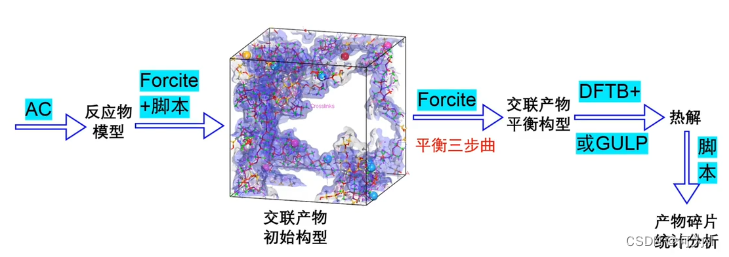
首先建模,使用AC模块将反应物放到一起构成反应模型,
平衡三部曲:几何优化+退火+分子动力学模拟。
案例五、表面活性剂对油-水界面性质影响的DPD模拟
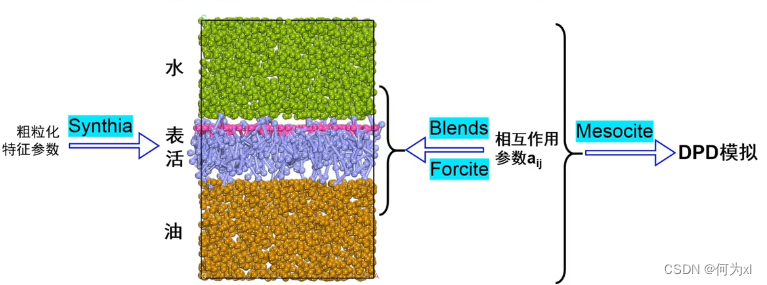
首先我们可以使用模块 Synthia 来表活分子的长链部分来计算一个参数——粗粒化特征参数。然后可以建立表活分子的珠子模型。然后通过Blends 和 Forcite 这些来计算 这些珠子之间的相互作用参数;在使用Mesocite 进行 DPD模拟。


















 1069
1069

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











