







【Lammps】lammps常用入门建模命令
前言
本文中,采用对 Ar 原子体系运动的模拟
模拟思路:

脚本总览
# 2d Lennard-Jones gas
#---------------Initialize Simulation -------------------------
units lj
dimension 2
boundary p p p
atom_style atomic
#-------------- Create Atoms Intial Conditions----------------
lattice hex 1.0
region mybox block 0 20 0 10 -0.1 0.1
create_box 1 mybox
region 2 block 0 10 0 10 -0.1 0.1
create_atoms 1 region 2
mass 1 1.0
velocity all create 2.5 87287
#---------------- Define Interatomic Potential ----------------
pair_style lj/cut 2.5
pair_coeff 1 1 1.0 1.0 2.5
neighbor 0.3 bin
neigh_modify every 20 delay 0 check no
fix 1 all nvt temp 0.5 0.5 0.01
fix 2 all enforce2d
#--------------- Run MD Simulation ----------------------------
dump 1 all custom 100 toEquil.lammpstrj id type x y z vx vy vz
thermo 500
run 10000

1、units
①主要作用:定义单位类型。units命令用来定义模拟过程中使用的单位类型,它决定了所有输入脚本、数据文件和所有输出到屏幕、日志文件以及dump文件中物理量的单位。
②语法:units style;
style 可取 lj or real or metal or si or cgs or electron or micro or nano;金属中一般用metal就行,不用做过多改变。
2、dimension
dimension命令用于定义模拟的维度。在LAMMPS中,默认情况下运行3D模拟。要运行2D模拟,需要在设置仿真盒子之前使用此命令。重启文件也存储此设置 。
在本例中使用 二维,故需要设置。同时进行二维模拟时,设置空间大小可以使 Z 轴的数值设为 -0.1 0.1.
3、boundary
①设置模拟box的边界条件
②语法:boundary x y z
boundary命令设置box的边界条件;参数xyz分别设置该维度的边界条件,最基本的边界条件有四种:p、f、s、m;
-
p是周期性边界:当粒子从一侧飞出,会从相对应的另一侧再次进入box,一般情况下不会发生“lost atoms”丢失原子错误;
-
f是固定边界:参数f表示box的表面在该方向上被固定住,不因体系压力的变化发生移动,如果原子从一个表面飞出,会提示“lost atoms”错误,设置thermo_modify lost可以允许原子丢失,否则模拟过程会被终止;
-
s是收缩性边界条件:参数s表示在该方向上,盒子的表面会根据体系的压力大小或原子运动情况发生移动。一般情况下,盒子会配合原子的移动,尽量把原子包含在box内,但是当原子速度过快,位移太大时原子也可能会飞出盒子,造成“lost
atoms”错误。 -
m:非周期最小值包覆边界条件
备注:lammps支持对同一方向的两个边界设置不同的边界条件,对y方向下表面设置f边界、上表面设置s边界,可以写为:boundary p fs p。
4、atom_style
① atom_style命令用来定义模拟过程中原子的类型
② 语法:atom_style style args
-
style 为amoeba or angle or atomic or body or bond or charge or dipole or dpd or edpd or electron or ellipsoid or full or line or mdpd or molecular or oxdna or peri or smd or sph or sphere or or bpm/sphere or spin or tdpd or tri or template or hybrid
-
atom_style命令用来定义模拟过程中原子的类型。我们模拟的基本构成是原子。不同的对象粒子所具有的属性是不同的。不同粒子所具有的属性是不同的,对于某个粒子对象,粒子不存在的属性就不需要设置,以便节省内存,加速运算;当style为full时,其属性包括:粒子编号,粒子所属分子的编号,粒子类别(atom type),带电量,x坐标,y坐标,z坐标。但如果你的粒子是金属粒子,比如Fe,那么粒子(style为atomic)的属性就是:粒子编号,粒子类别,x坐标,y坐标,z坐标。
研究范围是金属方向,所以一般为:atom_style atomic。
原子模型构建

5、lattice
定义一个供其它命令使用的晶格。
- 语法:lattice style scale keyword values …
- style = none or sc or bcc or fcc or hcp or diamond or sq or sq2 or hex or custom;
- scale = scale factor between lattice and simulation box;对于除了LJ units外的所有其他单元,scale是以距离为单位的晶格常数(lattice constant in distance units).
- keyword=origin or orient or spacing or a1 or a2 or a3 or bias.
- 介绍
在LAMMPS中,晶格仅仅是空间中点的集合,由一个具有基原子的单元细胞决定,该单元细胞在所有维度中无限被复制。lattice命令可以用来定义各式各样的晶格。
lattice在lammps以两种方式被应用,第一种是利用create_atoms命令在模拟框的晶格格点上创建原子,create_atom命令是给晶格格点上的基原子分配不同的原子类型;第二种是被create_box、region、velocity等命令用作距离单位。
lattice命令需要与模拟维度保持一致,style sc or bcc or fcc or hcp or diamond被用作3D问题,而style sq or sq2 or hex 被用作2D问题。
Eg:lattice fcc 4.05 表示晶格类型是fcc,晶格常数为4.05,单位是埃米。
6、region
region命令用于定义一个空间几何区域。
语法:region ID style args keyword arg ...
ID:用户赋予的名字;这个区域的名字。
style:delete or block or cone or cylinder or ellipsoid or plane or prism or sphere or union or intersect
delete:没有参数;block: xlo xhi ylo yhi zlo zhi是三个维度上的最小值和最大值;
cone:dim c1 c2 radlo radhi lo hi。
举例如下:
powershell lattice fcc 4.05 region box1 block 0 30 0 30 0 30
意思是创建一个名字为box1的block,其中x,y,z方向上的长度均为30倍的晶格常数。注意:这里的30是30倍的意思,实际长度为30乘以4.05埃米,如果想要让30代表实际长度,既让其单位为埃米,则只需要在后面加上units
box即可,如下:
powershell region box1 block 0 30 0 30 0 30 units box
在本示例中,创建一个名为 mybox 的block(几何空间),x,y 方向上的长度均为20倍的晶格常数,由于是在二维平面进行模拟,故设置 z取 -0.1,0.1。
lattice hex 1.0
region mybox block 0 20 0 10 -0.1 0.1
7、create_box
lammps 模拟时所有原子都必须在一个有限的区域内,这个有限的区域就是box,使用create_box命令创建。
该命令还固定了该模拟区域中有几类原子;
powershell create_box N region-ID keyword value ...
N:本次模拟使用的原子种类数;region-ID:用来模拟的区域的ID;keyword = bond/types or angle/types or dihedral/types or improper/types or extra/bond/per/atom
or extra/angle/per/atom or extra/dihedral/per/atom or
extra/improper/per/atom or extra/special/per/atom
create_box 1 mybox
对于,region和box,可以这样理解:
在LAMMPS中,region命令用于设定具有一定尺寸的区域,该区域的尺寸确定之后,就可以在这个区域内创建盒子。而create_box命令则是利用region命令所创建的区域,在该区域内创建盒子 。
8、creat_atoms
create_atoms命令是lammps建模命令中必不可少的一个命令,主要功能是在指定区域内填充一定数量的原子。
create_atoms命令格式为:
create_atoms type style args keyword values ...
其中,type指定填充原子的类型,style指定填充原子的方法,lammps提供了box 、region 、single和random 4种填充原子方式。
1. 在盒子box内填充原子
create_atoms type box
box类型是最简单的一种用法,在模拟盒子内按照指定的晶格类型填充指定类型的原子,执行这句命令之前需要提前定义box和lattice。
create_atoms 1 box
命令执行结果如下图所示:

2.在指定区域(region)内填充原子
create_atoms type region region-ID
对于复杂结构体系或者不同形状晶体结构,可以配合region命令实现分区域填充原子。
lattice fcc 3.61
region box block -10 10 -10 10 -10 10
create_box 1 box
region Cu_sphere sphere 0 0 0 5
create_atoms 1 region Cu_sphere
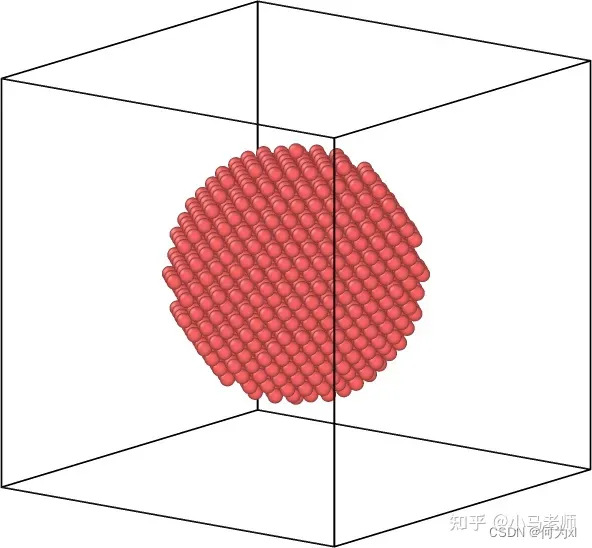
3.生成单个原子
create_atoms type single x y z
create_atoms可以在指定的坐标点上生成单个原子
lattice fcc 2.86
region box block 0 20 0 20 0 20
create_box 1 box
create_atoms 1 single 10 10 10
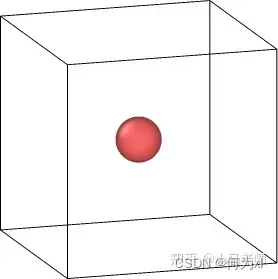
4.随机生成原子
create_atoms type random N seed region-ID
random类型在指定区域内随机生成原子,N为生成原子总数,seed为随机数种子,region-ID指定填充原子区域。
lattice fcc 2.86
region box block 0 20 0 20 0 40
create_box 1 box
region fe block 0 20 0 20 5 35
create_atoms 1 random 30 8763 fe
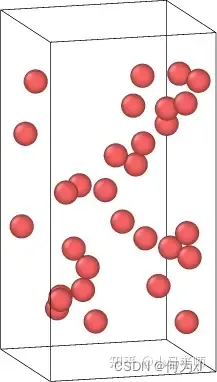
region 2 block 0 10 0 10 -0.1 0.1
create_atoms 1 region 2
mass 1 1.0
velocity all create 2.5 87287
9、mass
mass是LAMMPS中的一个命令,用于设置分子的质量。这个命令通常在输入文件的开始部分使用,用于为模拟设置合适的参数。
mass命令的语法如下:
mass <id> <value>
其中,表示原子或分子的ID,可以是单个原子或多个原子/分子的名称,也可以是一个数字索引。表示要设置的质量值,单位是原子质量单位(amu)。
例如,要设置一个名为"atom1"的原子的质量为12.000000 amu,可以使用以下命令:
mass atom1 12.000000
如果要设置多个原子的质量,可以使用逗号分隔它们的ID和质量值,例如:
mass atom1 12.000000 mass2 1.008 atom3 33.973
这里,我们设置了第一个原子的质量为12.000000 amu,第二个原子的质量为1.008 amu,第三个原子的质量为33.973 amu。
如果要设置整个分子的质量,可以使用通配符"*"来匹配所有原子/分子的名称,例如:
mass * 12.000000
这里,我们设置了所有原子/分子的质量为12.000000 amu。
需要注意的是,mass命令只是设置了分子的质量,而没有进行任何计算。如果需要进行力的计算,还需要使用其他命令,如pair_style、pair_coeff等。
10、velocity
在LAMMPS中,使用velocity命令设置原子的速度。
command format为:
velocity group-ID style args keyword value …
其中,group-ID指定哪一组原子将要被设置速度,style决定了速度设置的类型。 常见的速度设置方式有以下三种:
(1)velocity create进行初始化。分子动力学模拟需要有一个初始的状态,因此,当原子模型建立完成,进行minimize能量最小化之后,需要进行速度初始化,体系的温度是由原子的温度计算得到,因此,这个过程也称为温度初始化。使用的命令为 velocity all create 300 4928459;
velocity all create 300 4928459
这一条语句的作用是对所有原子进行速度初始化,总体系的温度为300k,4928459为随机数,每个原子的速度被随机设置,默认的情况下,原子的速度符合正态分布。
也可以设置dist参数,使原子的速度符合高斯分布:
velocity all create 300 4928459 dist gaussian
当然,也可以对部分原子进行速度初始化,只需要改变原子组即可:
velocity mobile create 298 4928459
(2)velocity set设置原子在特定方向上的速度
在拉伸、剪切等动力学模拟中,需要这种一部分原子沿着某一个方向移动,可以使用velocity set命令设定特定方向的速度。
例如在沿着Z轴进行拉伸模拟时,需要把底部原子固定住,使上部(top)原子沿着Z轴施加一个速度,可以写为:
velocity top set NULL NULL 2 sum yes units box
上句代码中 NULL NULL 2 表示不设置x和y方向的速度,仅设置z方向速度,sum yes的意思是将速度2加到原子当前时刻速度分量上,如果不写sum yes,则表示忽略原来的速度,直接将z方向速度直接设为2。
默认的单位为晶格单位(lattice),units box关键字可将单位设置为实际速度单位,具体单位与体系units有关。
也可以多个方向同时设置:
velocity top set 2 2 0 sum yes
将x、y方向移动速度设为2(晶格单位),z方向速度为0,覆盖掉原来的速度值。设置之后相当于top组原子沿着x和y方向的对角线方向移动。
(3)velocity ramp设置速度场
lammps允许在某一个方向上按照不同的坐标位置设置不同的速度,例如:
velocity flow ramp vx 0.0 5.0 y 5 15
这句代码是将y坐标从5到15的原子在x方向的速度设为0到5,y坐标小于5的原子x方向速度为0,y坐标大于15的原子x方向的速度为5,中间的原子x方向速度按照比例关系进行设定,例如y坐标为10的原子x方向上的速度为2.5。
模拟参数的设置
设置作用势
颗粒之间相互作用模型

在分子动力学模拟中, 粒子间的相互作用是通过势函数命令来指定的。 一般地, 原
子间的势函数命令有: pair_style,pair_coeff 和 pair_modify。 而对于分子内的原子间
的相互作用还需要:bond_style 和 bond_coeff 命令来确定, 这里介绍三个势函数命令
即: pair_style,pair_coeff 和 pair_modify。
10、pair_style
( 1) pair_style 命令设置了每对原子截断半径内的相互作用的势函数类型。 这种势
函数所定义的原子间的相互作用是随时间变化的,与此对应的 bond_style 所设
置的原子间的相互作用一般不随时间变化。 在 LAMMPS 中, 一些多体势也归
类为对势, 例如 EAM,Stillinger-Weber,Tersoff,REBO 等。 因为这些势函数所定
义的原子间相互作用也是随时间变化的。 使用 hybrid 类型的势函数可以定义
不同类型的原子以不同的势函数相互作用。 势函数的具体类型确定后, 不同类
型原子的势函数的具体参数由 pair_coeff 命令设置,也可以由 read_data 命令和
read_restart 命令进行文件读入。
pari_style 命令一般会设置一个全局的截断半径, 全局的截断半径是可以被
pair_coeff 命令中的截断半径重新覆盖的。 一般地, 在一个 in 文件里重新使用
pair_style 会抹去之前设置的所有 pair_style 和 pair_coeff 命令, 除了对于
pair_styletable 和 hybrid 的设置。
以下是所有 pair_style 可选类型的概述:
pair_style none:关闭势函数相互作用。
pair_style hybrid:多种类型的势函数。
pair_style hybrid/overlay: 多种可叠加的势函数。
pair_style beck: Beck 势, 最初用于模拟氦气。
pair_style body:体类型粒子间的相互作用。
pair_style bop: 基于量子力学计算 sigma 和 pi 键的键序势。
pair_style comb: 优化电荷作用的多体势。
pair_style coul/cut:截断库伦势。
pair_style dpd:耗散粒子动力学。
pair_style dsmc:直接模拟蒙特卡洛方法计算粒子碰撞。
pair_style eam: 嵌入原子势。
pair_style eim: 嵌入离子势。
pair_style gauss: 势函数的表达式具有 Gaussian 函数的形式。
pair_gayberne: 考虑各向异性的 LJ 势。
pair_style kim: KIM 项目势函数接口。
pair_style lcbop:长程键序势函数。
pair_style line/lj:线段类型粒子的 LJ 势函数。
pair_style lj/charmm:CHARMM 力场势函数。
pair_style lj/class2: COMPASS 力场势函数。
pair_style lj/cut:截断的 LJ 势
pair_style lj/expand: 扩展 LJ 势函数, 粒子半径可以为变量。
pair_style lj/gromacs: GROMACS 型 LJ 势。
pair_style lj/long: 长程 LJ 势。
pair_coeff
pair_coeff 命令指定了不同类型原子间势函数的具体参数。 其中 I,J 代表原
子类型, 可以利用具体的代表原子类型的数字表示, 要求 I<=J, IJ 间的相互
作用和 JI 间的相互作用是相同的。
采用星号可以同时表示多种原子类型。 此时 I,J 可以有四种表示形式:
,n,n,mn。 单个星号表示所有的原子类型 1 到 N。 一次类推,n 代表 1 到 n。
n代表 n 到 N。 mn 代表 m 到 n。
如果对同一对原子类型间设置多次势函数参数,新的参数会重新覆盖原来
的参数。 例如如下两行命令:
pair_coeff**101025
pair_coeff232010112
对于某些势函数当 I 不等于 J 的时候, 势函数的参数需要根据 I 和 I 之间或者
J 与 J 之间的参数进行计算。 此时可以通过 pair_modify 命令进行设置。 对于大
多数多体势来说, 势函数的参数是通过文件的形式输入到模拟中的, 此时需要
把用到的势函数文件放入相应的文件夹或指定势函数文件的路径。
( 3) pair_modify
pair_modify keyword values
其中 keyword 可选的参数有:pairshiftmixtabletable/disptabinnertabinner/disptailcompute
Pair_model 命令可以修改当前定义的势函数参数。
pair 关键字用于 hybrid 和 hybrid/overlay 两种类型的势函数。 利用该关键字可以修
改子势函数类型中的参数。 special 关键字和 pair 连用, 可以定义子类型势函数中的独
特的势函数参数。
pair_modifypairlj/cut/coul/long1speciallj/coul000000
mix 关键字用于当 I 不等于 J 时势函数参数的计算方法定义。 并不是所有的势函数
类型, 都可以使用 mix 中的计算规则进行不同原子间的势函数参数计算。 以下是 mix
参数的定义:
mixgeometric
epsilon_ij=sqrt(epsilon_iepsilon_j)
sigma_ij=sqrt(sigma_isigma_j)
mixarithmetic
epsilon_ij=sqrt(epsilon_iepsilon_j)
sigma_ij=(sigma_i+sigma_j)/2
mixsixthpower
epsilon_ij=(2sqrt(epsilon_i*epsilon_j)*sigma_i3*sigma_j3)/(sigma_i6+sigma_j6)
sigma_ij=((sigma_i6+sigma_j6)/2)^(1/6)
shift 关键字会影响在热力学参数输出中是否进行截断半径外的势能补偿。 但该命
令不影响模拟过程中实际力和轨迹的计算, 只是在最终势能或总能量输出的过程中,
补偿截断半径外的势能。
table 和 table/disp 关键字可以提前建立长程库伦力或长程耗散力的表格。在模拟时,
粒子间关于长程库伦作用项和长程耗散项的数值可以直接从表格中读取, 从而增加模
拟的效率。 可以通过查 manual 文件中确定哪些势函数可以建立这些表格。
tabinner 和 tabinner/disp 关键字可以设置上述表格中有关长程库伦力或长程耗散力
的截断半径。
tail 关键字为某些势函数的长程范德华力进行尾部校正。 这些校正一般不会影响原
子的动力学运动, 除非系统在恒压模式下模拟。 这些尾部校正也会影响到热力学量的
输出。尾部校正项是在一次 run 的开始统计原子数量并进行计算的。如果在模拟过程中,
原子有增加或丢失, 需要重新设置 run 进行校正计算。
pair_style lj/cut 2.5
pair_coeff 1 1 1.0 1.0 2.5
neighbor 0.3 bin
neigh_modify every 20 delay 0 check no
fix 1 all nvt temp 0.5 0.5 0.01
fix 2 all enforce2d
这段命令是用于设置分子间相互作用类型、截断距离和系数的。具体来说:
pair_style lj/cut 2.5:表示使用长程Lennard-Jones势和截断距离为2.5埃的相互作用类型。
pair_coeff 1 1 1.0 1.0 2.5:表示在lj/cut类型的相互作用中,将第一个原子之间的库伦势系数、第二个原子之间的库伦势系数、第三个原子之间的库伦势系数都设置为1.0,第四个原子之间的库伦势系数设置为2.5。
因此,这个命令的意思是:使用长程Lennard-Jones势和截断距离为2.5埃的相互作用类型,并设置第四个原子之间的库伦势系数为2.5。这样,LAMMPS在计算分子间相互作用时就会考虑这种类型的相互作用,并且根据指定的系数计算它们的强度。
计算近邻列表
neighbor
根据分子动力学原理我们需要计算每个原子的受力。而任何一个粒子的受力都来自于其周围粒子给它的合力。但是,我们知道只有距离较近的粒子才会对该粒子产生受力,距离较远的粒子给与的力可以忽略不计。这就是邻居的含义。所以要计算一个粒子所受的合力,首先要找到该粒子周围有哪些邻居,即建立邻居列表。每一个粒子都有自己的邻居列表。neighbor的语法是
neighbor <skin> <style> #举例:neighbor 3 bin
所谓邻居就是距离中心粒子一定范围内的粒子,这个范围称之为作用力的截断半径(r_c)。也就是以中心粒子为球心,r_c为半径内的所有粒子都是邻居。skin是比r_c多出来的一部分,所以建立邻居列表的时候是以r_c+skin为半径进行寻找的。为什么要这么做呢?这是因为建立邻居列表是挺费时间的一件事,那么好不容易建立起来那就尽可能多用几次。而事实上由于分子动力学模拟的时间步长较短,粒子在一个时间步长内走不了多少距离。那么只要邻居列表中的原子移动距离都小于skin,那么这个列表就可以在下一步中继续使用。这就是skin的含义。后面style指的是建立邻居列表的方法,最常用的就是bin方法。这个方法的具体含义可以查看分子动力学教材。入门只需要知道bin在大多数情况下都是最快的,用它就没错了。而r_c在后面的命令中设置。
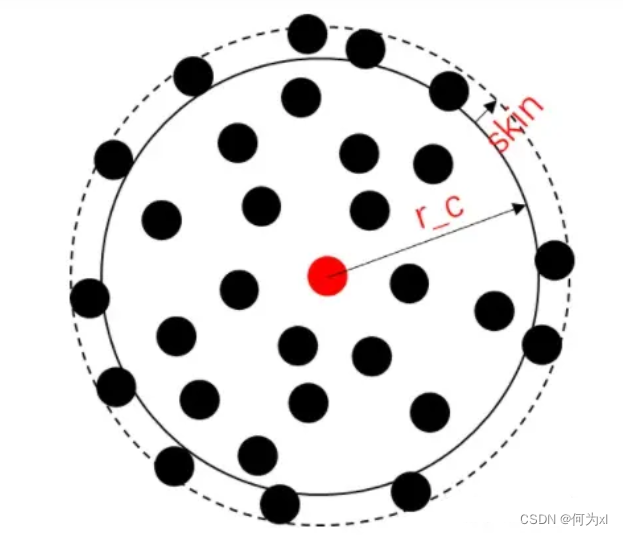
neigh_modify
neigh_modify命令设置邻居列表的更新频率。LAMMPS提供了很多方式设置邻居列表的更新频率。如果邻居中的粒子已经不在r_c+skin范围内了,那么邻居列表就必须要更新了。当需要更新而未更新时就会出现Dangerous builds。当算完之后LAMMPS输出的Dangerous builds不为零,那么你就要修改neigh_modify命令了,或者加大skin的值。这条命令使用比较复杂,需要根据模拟经验和具体的模拟测试确定最优值。对于新手,可以直接使用以下设置:
neigh_modify every 1 delay 0 check yes
以上都是计算设置,每个模拟这些设置都大同小异。下面开始就是针对模拟对象的设置。

15、fix

fix ID group-ID style_name keyword value ...
ID = user-assigned name for the fixgroup-ID = ID of the group of atoms to apply the fix tostyle = one of a long list of possible style names (see below)
nvt or npt or nphargs = arguments used by a particular style
举例如下:
fix 1 all npt temp 300 300 0.1 iso 0 0 1 drag 1
1是该fix命令的ID;all意思是对所有原子施加后面的条件;npt是等温等压系综;temp是设置温度的关键字,temp 300 800 0.1命令中,temp后的第一个参数300是初始温度,第二个参数300是截至温度,第三个参数0.1是温度的阻尼系数,设置它的意思是使其在升温或者降温过程中不会出现太大的波动,一般来说阻尼系数是100倍的tempstep。iso是控压的,因为设置的是npt系综,所以温度和压力必须设置,iso是控压方式,与之对应的还有aniso,其中iso是对x,y,z三个方向进行联合控压,或者说是耦合控压,在控压时要改变模拟盒子的大小,x,y,z三个方向同时改变。与之相对的是aniso,是非耦合控压,x,y,z三个方向不是同时改变。iso后跟的是初始压强,结束压强,和阻尼系数(为了使压强在增加或者降低过程中不会出现太大波动)。
动力学模拟

17、dump
dump ID group-ID style N file attribute1 attribute2 ...
ID = user-assigned name for the dump
group-ID = ID of the group of atoms to be dumpedstyle = atom or atom/adios or atom/gz or atom/zstd or atom/mpiio or cfg or cfg/gz or cfg/zstd or cfg/mpiio or cfg/uef or custom or custom/gz or custom/zstd or custom/mpiio or custom/adios or dcd or grid or grid/vtk or h5md or image or local or local/gz or local/zstd or molfile or movie or netcdf or netcdf/mpiio or vtk or xtc or xyz or xyz/gz or xyz/zstd or xyz/mpiio or yaml.
N = dump on timesteps which are multiples of N
file = name of file to write dump info
toattribute1,attribute2,… = list of attributes for a particular style
举例如下:
dump 1 all custom 1000 Al.xyz type x y z
其中1是用户指定的dump名字,all是关键字,意思对所有的原子施加后面的操作,custom是自定义输出,1000的意思是每1000步输出一次,输出到后面的Al.xyz文件中,该文件中输出的信息有type(原子类型),和x,y,z坐标。
整个过程中的所有信息保存在log.lammps文件中。
16、thermo 和 thermo_style
设置输出信息/格式。
thermo N
N = output thermodynamics every N timesteps
thermo_style style args
style = one or multi or yaml or customargs = list of arguments for a particular style
举例如下:
thermo 1000
thermo_style custom step lx ly lz press pxx pyy pzz pe temp
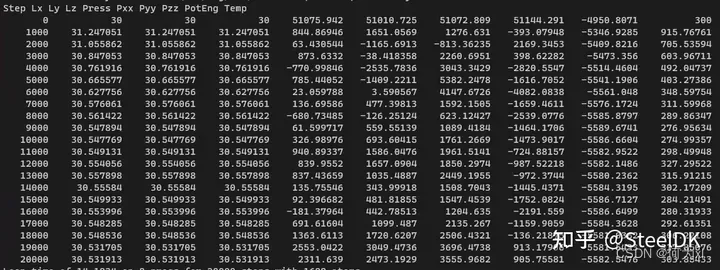
thermo 1000意思是每1000步在屏幕上输出一次。thermo_style后面是在屏幕上输出的信息。custom命令后的内容就是在屏幕上输出的,可以看到输出的运行步数step,三个方向上的长度lx,ly,lz,模拟体系的压力press,三个方向上的压力pxx,pyy,pzz,pe是模拟体系的势能,temp是模拟体系的温度。下图中即显示了上述信息,可以看到最后一行是温度,在模拟开始时温度为300k,由于前面fix命令中设置了temp 300 300 0.1,所以截至温度也是300K,中间会有升温和降温过程,如果最终温度没到300K,则可能是运行的步数不够。
run
在LAMMPS中,使用run命令可以设定运算步数并启动运算。具体来说,run命令的语法如下:
run <steps>
其中,表示要运行的步数。例如,要运行10000步,可以使用以下命令:
run 10000
此外,run命令还有其他高级应用。例如,可以使用以下命令来运行到特定的步数:
run 1000 upto 5000
这将从第1000步运行到第5000步。还可以使用以下命令来运行每隔N步执行一次某个命令:
run 1000 every 100 command
这将从第1000步开始,每隔100步执行一次command命令 。
参考资料:
https://zhuanlan.zhihu.com/p/349566666?utm_id=0

















 887
887











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











