






(1)使用Mash创建每个基因组的Sketches
注:mash要在funannotate下才能使用,所以要先conda激活funannotate哦~
(mash_sketch.sh内容)
#!/bin/bash
# 使用Mash创建每个基因组的Sketches
for genome in *_genomic.fasta;
do
mash sketch -m 2 -k 21 $genome
done#然后手动将所有的.mash文件放到同一目录下
(2)使用Mash比较所有Sketches,并输出距离矩阵
$ mash dist *.msh > distances.tsv
# 使用Mash比较所有Sketches,并输出距离矩阵
$ echo "" # 输出距离矩阵
echo "Mash距离矩阵:"
$ cat distances.tsv #获得遗传距离表格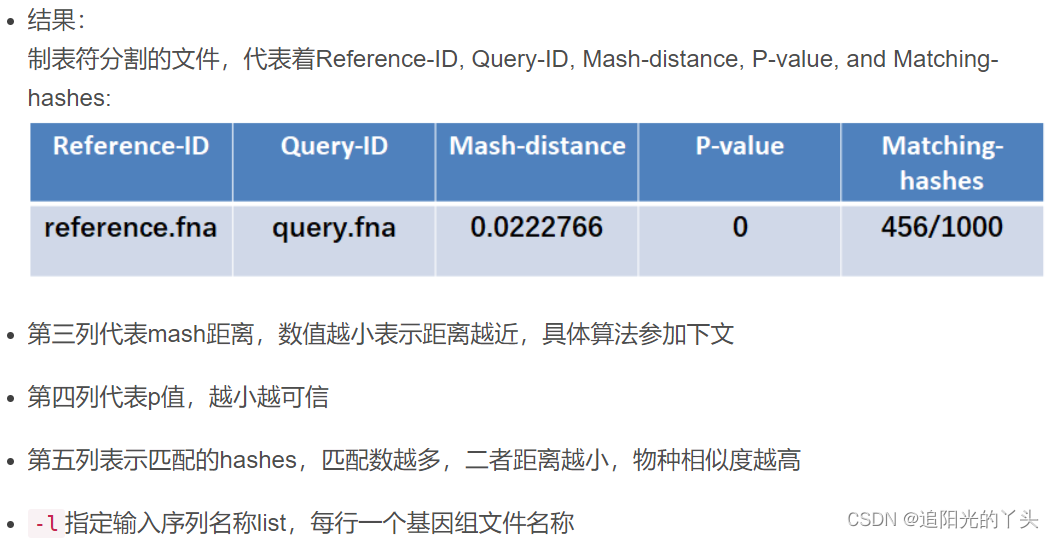
(3)使用mashtree软件去建树
$ conda install -c anaconda mashtree
#安装mashtree软件去进行建树
#好像不需要激活直接就可以使用
$ mashtree *.msh > tree.dnd
#利用生成的.mash文件进行建树,得到树之后下载到本地,用iTOL打开,只能看到拓扑关系,无法显示自展值#这是根据遗传距离建的树,体现遗传距离上的远近
#老师说需要用单拷贝基因构建系统发育树,那样才是体现系统发育关系,更准确鉴定物种


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









