






一、用R做花瓣图
注!!!先把前面数据都导入进去之后再生成花瓣图,现在已经更新了3、4、5三个版本,注意文件名的对应,然后导出的时候选择PDF,选择A4格式导出
#做花瓣Venn图,目的是统计一下现在得到的正选择结果有多少重复的和特异性的,用R做
#设置工作目录
setwd(dir = "C:/Users/Lenovo/Desktop/repeat")
# 导入所需的包
library(grid)
library(futile.logger)
library(VennDiagram)
library(venn)
library(ggplot2)
# 读取文件中的基因列表
#我这次有5个文件,引号中是文件名,就是先把这些数据导入进去
genes1 <- read.table("branchsite_all.txt", header = FALSE, col.names = "Gene")
genes2 <- read.table("site_m1m2_all.txt", header = FALSE, col.names = "Gene")
genes3 <- read.table("site_m7m8_1368.txt", header = FALSE, col.names = "Gene")
genes4 <- read.table("site_m7m8_all.txt", header = FALSE, col.names = "Gene")
genes5 <- read.table("w999_quickevolution.txt", header = FALSE, col.names = "Gene")
# 将基因列表转换为字符向量,这样我5个文件就转换成了5个字符向量,下面就用genesX代替
genes1 <- as.character(genes1$Gene)
genes2 <- as.character(genes2$Gene)
genes3 <- as.character(genes3$Gene)
genes4 <- as.character(genes4$Gene)
genes5 <- as.character(genes5$Gene)
# 生成花瓣图,这个会分别计算5个中两两之间、三个、四个、五个的重叠度,注意category那里要填写一下文件名称,其他地方无需改动
venn.plot <- draw.quintuple.venn(
area1 = length(genes1),
area2 = length(genes2),
area3 = length(genes3),
area4 = length(genes4),
area5 = length(genes5),
n12 = length(intersect(genes1, genes2)),
n13 = length(intersect(genes1, genes3)),
n14 = length(intersect(genes1, genes4)),
n15 = length(intersect(genes1, genes5)),
n23 = length(intersect(genes2, genes3)),
n24 = length(intersect(genes2, genes4)),
n25 = length(intersect(genes2, genes5)),
n34 = length(intersect(genes3, genes4)),
n35 = length(intersect(genes3, genes5)),
n45 = length(intersect(genes4, genes5)),
n123 = length(intersect(intersect(genes1, genes2), genes3)),
n124 = length(intersect(intersect(genes1, genes2), genes4)),
n125 = length(intersect(intersect(genes1, genes2), genes5)),
n134 = length(intersect(intersect(genes1, genes3), genes4)),
n135 = length(intersect(intersect(genes1, genes3), genes5)),
n145 = length(intersect(intersect(genes1, genes4), genes5)),
n234 = length(intersect(intersect(genes2, genes3), genes4)),
n235 = length(intersect(intersect(genes2, genes3), genes5)),
n245 = length(intersect(intersect(genes2, genes4), genes5)),
n345 = length(intersect(intersect(genes3, genes4), genes5)),
n1234 = length(intersect(intersect(intersect(genes1, genes2), genes3), genes4)),
n1235 = length(intersect(intersect(intersect(genes1, genes2), genes3), genes5)),
n1245 = length(intersect(intersect(intersect(genes1, genes2), genes4), genes5)),
n1345 = length(intersect(intersect(intersect(genes1, genes3), genes4), genes5)),
n2345 = length(intersect(intersect(intersect(genes2, genes3), genes4), genes5)),
n12345 = length(intersect(intersect(intersect(intersect(genes1, genes2), genes3), genes4), genes5)),
category = c("branchsite_all.txt", "site_m1m2_all.txt", "site_m7m8_1368.txt", "site_m7m8_all.txt", "w999_quickevolution.txt"),
fill = c("skyblue", "pink", "lightgreen", "orange", "purple"), # 添加新颜色选项
lty = "blank",
cex = 1.2,
fontface = "bold",
fontfamily = "sans"
)
###以下是生成3个文件的花瓣图的代码,下面的genes1,2,4是指上文中1,2,4文件,这个都可以自己定义
# 生成花瓣图
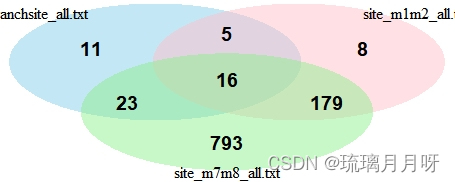
venn.plot <- draw.triple.venn(
area1 = length(genes1),
area2 = length(genes2),
area3 = length(genes4),
n12 = length(intersect(genes1, genes2)),
n23 = length(intersect(genes2, genes4)),
n13 = length(intersect(genes1, genes4)),
n123 = length(intersect(intersect(genes1, genes2), genes4)),
category = c("branchsite_all.txt", "site_m1m2_all.txt", "site_m7m8_all.txt"),
fill = c("skyblue", "pink", "lightgreen"),
lty = "blank",
cex = 1.2,
fontface = "bold",
fontfamily = "sans"
)
二、在线网站做upset图
因为搜索过后,感觉维恩图最多是7个,没有8个的,所以就又学了这个
进入网站,准备好数据,直接提交生成图片就行
数据大概是这个样子,每个物种一列
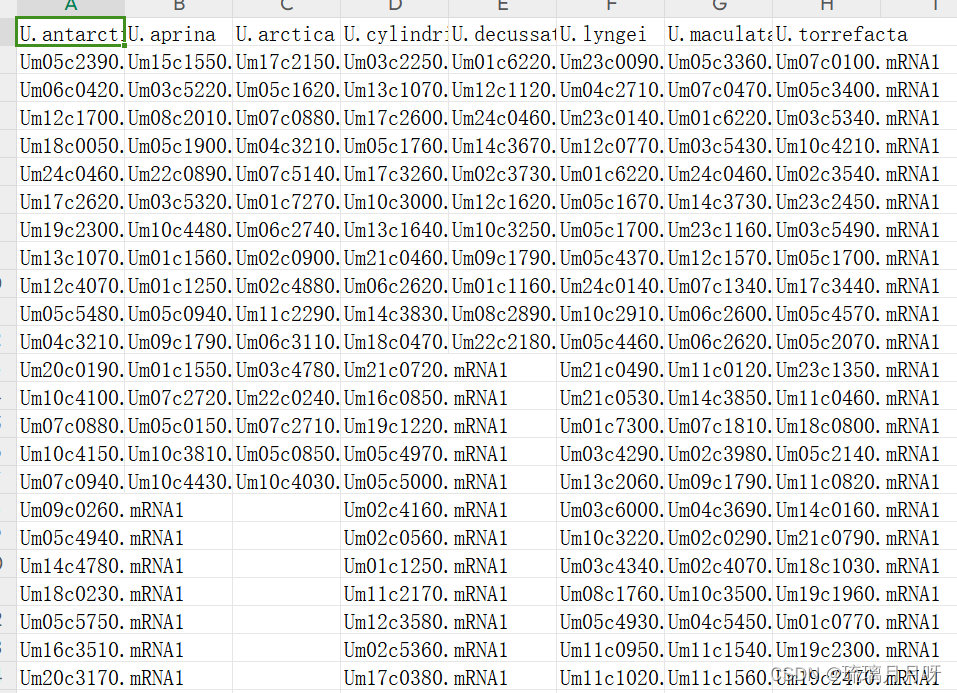
提交之后稍等生成图片这样
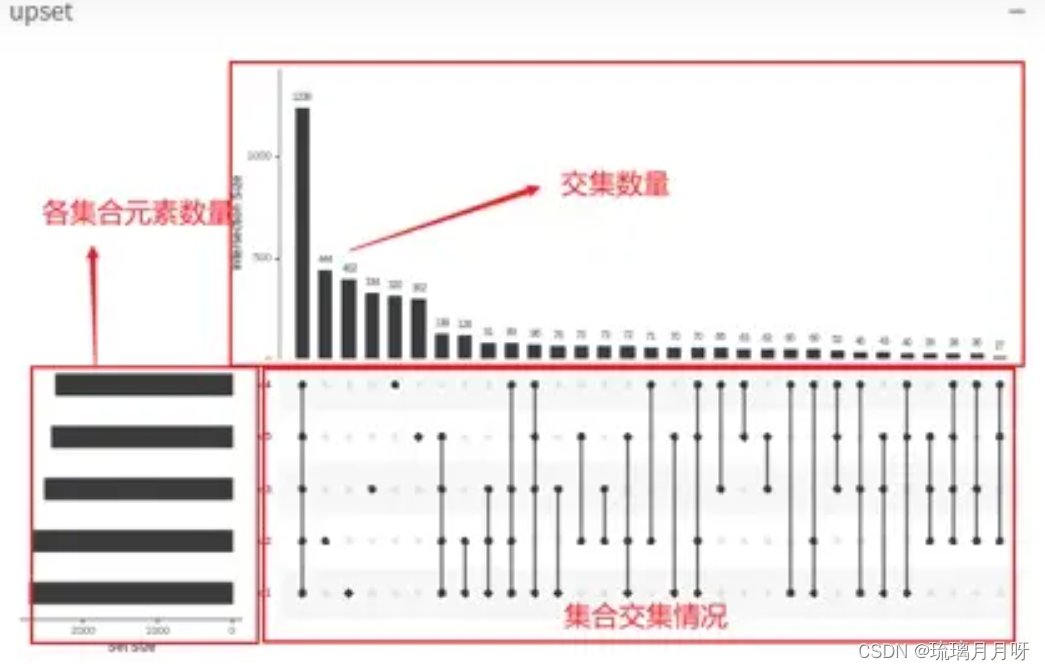
左边是物种,及每个物种里有多少个输入项
上方表示重合的数量
下方单个点表示是自己单独的个数,连成线表示XX和XX是有重合的,重合的数目是上方的柱状数
(只能说这种展现方式好聪明哦~棒棒哒!)
导出的时候可以选择一个png一个pdf,一个留着先看,另一个留着美化修饰。
三、在线网站画花瓣图
输入数据,每个物种有多少基因个数,核心共有的有多少,然后生成图片就行

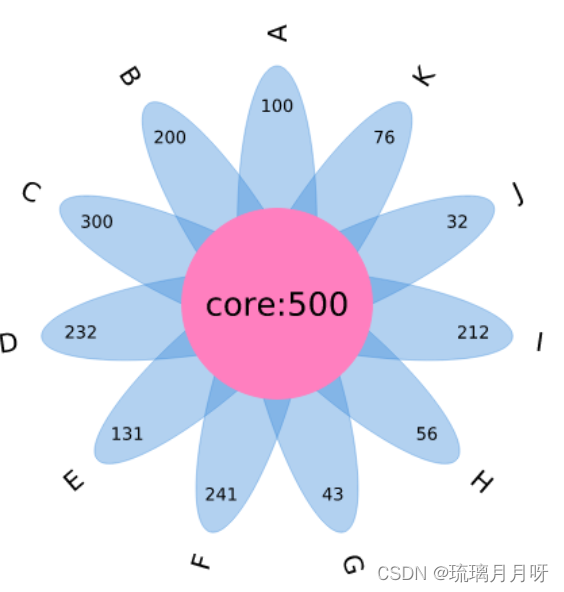
四、补充生信作图网站
BioLadder-生物信息在线分析可视化云平台 登录即用,免费
https://nmdc.cn/analyze 国家微生物科学数据中心(会有一些生信分析工具,像MUMer,tRNA-scan等,不做图)
https://analysis.mypathogen.org/workflow/ 微生物数据分析云平台(也是一些生信分析工具,像特异基因查找等,不做图)
这个只能做三维恩图,稍微有点拉跨
ChiPlot szq强推,有B站教程,比较好
微生信-在线生物信息学分析、可视化云平台 这个里面作图也蛮丰富的,免费















 1106
1106











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









