






PAML网站: http://abacus.gene.ucl.ac.uk/software/paml.html
!!!注1:如果想单独前台跑paml的话,那就准备好序列文件、树文件、控制文件,然后用codeml运行控制文件就行,如: codeml ctl_site_none
注2:在运行PAML之前先对获取到的gapfiltered目录下的文件进行假基因的鉴定,运行pseudogene_count.py 代码,命令如下:python3 pseudogene_count.py <工作目录>
python3 ~/Qxy/qxyjiaoben/fromzyr/pseudogene_count.py ~/Qxy/knowngenome/gongxianxing/0926bidui
补充一下疑惑:
1)site model、branch model 、branchsite model区别是什么呢
位点模型是序列不同碱基位点之间的差异,考虑的是整体水平,没有前景枝和背景枝的区分,所以可以用来分析石耳属整体对于低温的适应;
枝模型是不同分支之间的差别,有前景枝和背景枝之分,本课题暂未用到;
枝位点模型是在考虑前景枝和背景枝等不同分枝的基础之上,还考虑位点的差异性,所以可以用来分析南极石耳相较于其他石耳对于极端寒冷气候的适应。
一、B站教程 以鲸鱼为例 Branch test 枝模型计算dN/dS进化速率
但是我好像不用Branch model,我需要用的是site model和branch site model
1.1背景知识
dN/dS : The ratio of the rate of non-synonymous substitutions (dN) to the rate of synonymous substitutions (dS)
substitutions:氨基酸序列中的替换,有同义替换synonymous和非同义替换non-synonymous两种
data数据:divergence data 取物种中某一个体序列代表整个物种序列,不考虑多态性
synonymous同义替换:mostly natural 大多数是中性的,既没有正向选择也没有负向选择(因为虽然核苷酸变了,但是氨基酸没有发生改变,变里不变表)
non-synonymous非同义替换:mostly deleterious 大多数是吃亏的,会对个体产生一定不利的影响(氨基酸一变,蛋白质可能也就变了,结构功能等就都不一样了)
netural中性条件:dN/dS=1 positive selection:dN/dS > 1 negation selection:dN/dS < 1 该方法非常保守!!!一般蛋白质都不会轻易发生改变的
通常检测值是0.05 -0.4 少数超过1的例子:MHC
w是dN/dS的估计值,通过Maximum Likelihood最大似然法计算得到
零假设:所有枝系有共同的w备择假设:一些枝系有不同的w
最终根据计算得到的P值,来看选择哪个假设
1.2 准备文件 好像是一个基因去进行PAML分析
1)同源基因密码子序列比对结果文件Phylip格式
第一行是物种数目和密码子的数目
后面接着几行每行就是物种名字(30个字符以内)空几个空格(双空格表示物种的结束),跟着就是密码子序列

2)树文件,含拓扑结构,可以有根可以无根,也可带枝长信息
第一行是物种数目和1棵树,下面是树结构,树中物种名一定要和序列中相一致,且顺序对应
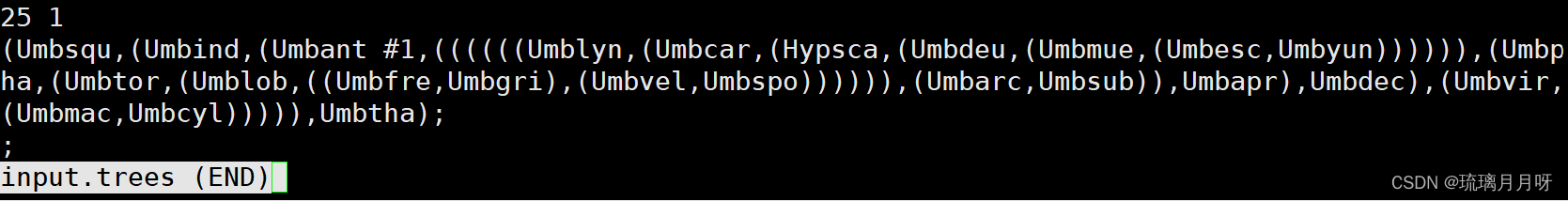
3)控制文件 codeml.ctl 执行 关键※是model=0 (进化树上所有分枝w一致)零假设 然后设置好seqfile treefile outfile路径
model=2 备择假设 w不一样,在树文件上用#1做标记,标#1的一支其w一致
1.3 结果解读
每次运行完代码会生成result.txt结果文件,打开,寻找其中的lnL似然率(在树的下面)
Null(lnL0):lnL(ntime:14 np:16): -1295.438022 +0.000000
Alternative(lnL1): lnL(ntime:14 np:17): -1292.210434 +0.000000
lnL1-lnL0 Chi^2 df=np1 -np0 满足卡方分布,也知道df,就可以算出P
【用R去进行计算,得到P为0.072,小于10%,显著,否定零假设,选择备择假设】
然后再去结果文件中找w值,零假设null: omega(dN/dS)=0.12795 备择假设 w(dN/dS)for branches = 0.08179 0.21097 有两个w值,因为当初设置树文件的时候标了个#1,就表示那一支是一个w,其他枝是一个w
二、自己数据运行Branchsite model
需要做:将gapfilter中的数据进行比对,然后RAXML建树 (4dtv的也行?)
运行师姐代码 需要先有树文件,树文件统一用一个就行,大差不差,后期运行site model时可能会根据基因的不同调整树的结构
2.1 输入文件批量准备paml_inputfile.py
注意!!!先看一下gapfiltered文件夹底下,别有啥杂七杂八的,影响代码运行!!!
#python3 paml_inputfile.py <参考序列文件名> <工作目录路径> <前景物种名称>(与文件中名字相对应,多个物种逗号分隔) <进化树文件路径> <模型>(branchsite、site)选一个
nohup python3 /ifs1/User/wuqi/Qxy/qxyjiaoben/fromzyr/paml_inputfile.py Umbilicaria_antarctica_genomic.fna /ifs1/User/wuqi/Qxy/knowngenome/gongxianxing/0828bidui Umbilicaria_antarctica_genomic /ifs1/User/wuqi/Qxy/knowngenome/gongxianxing/0828bidui/RAxML_bipartitions.output branchsite &运行结果是paml的输入文件,准备文件,很快速
2.2 控制文件批量准备
使用方法:python3 paml_ctl.py /path/to/workdir branchsite fix #注:工作目录下需要有PAML文件夹,且PAML目录下有相关文件
nohup python3 /ifs1/User/wuqi/Qxy/qxyjiaoben/fromzyr/paml_ctl.py /ifs1/User/wuqi/Qxy/knowngenome/gongxianxing/0828bidui branchsite fix &
nohup python3 /ifs1/User/wuqi/Qxy/qxyjiaoben/fromzyr/paml_ctl.py /ifs1/User/wuqi/Qxy/knowngenome/gongxianxing/0828bidui branchsite nofix & 注:fixoption指的是omega值,要分别计算一下零假设fix=1和备择假设 nofix=0,意思就是零假设认为大家进化速率完全一致,而备择假设认为前景背景不一致
运行结果是paml的控制文件,很快速
2.3 PAML脚本批量运行
paml_run.py
#是运行PAML的脚本,运行命名是:python run_paml_script.py <工作目录路径> <并发进程数>
nohup python3 /ifs1/User/wuqi/Qxy/qxyjiaoben/fromzyr/paml_run.py /ifs1/User/wuqi/Qxy/knowngenome/gongxianxing/0828bidui 20 &#注:工作目录下要有PAML文件夹
运行结果是paml的结果文件,这个运行时间近一周左右
2.4 卡方检验批量运行
python3 paml_chi2.py <参考基因组> <模型> <工作目录路径>
nohup python3 /ifs1/User/wuqi/Qxy/qxyjiaoben/fromzyr/paml_chi2.py Umbilicaria_antarctica_genomic.fna /ifs1/User/wuqi/Qxy/knowngenome/gongxianxing/0828bidui &#这是已经进行完PAML运算后,对其结果进行解读利用,就是看两个卡方值之间大小比较
运行结果是result文件,有统计好的omega值、P值等,还需要对P值进行FDR校正,做一下这个
2.5FDR校正
!!!补充:FDR(False Discovery Rate)是一种用于多重假设检验的校正方法,用于控制发现假阳性的概率。FDR之前需要先进行多重假设检验,比如t 检验、方差分析或卡方检验等。
so 我们上一步已经计算了两次假设的卡方值和P值,下面我们用Benjamini-Hochberg 方法(BH法)进行FDR校正。
哦哦哦哦!就是P value和Q value!!!梦幻联动了
将paml_result.txt结果传输到本地,用EXCEL操作
1)将所有基因的 p 值按照从小到大的顺序排列
2)计算每个基因 FDR 校正后的阈值:qi=pi*N/i 其中pi是第i个基因原始p值,N是总基因数,i是当前基因排名
3)对于每个基因,如果其原始 p 值小于等于对应的 FDR 校正阈值,则认为该基因通过了 FDR 校正,否则认为该基因未通过 FDR 校正。
4)最终得到通过 FDR 校正的基因集合,以及它们对应的 FDR 校正阈值(Q值)和原始 p 值
5)然后咱根据通过FDR检验的qvalue,选择显著性值(0.05或0.01)看是否通过显著性检验
6)可以把w=1中性突变给去掉,然后重做一遍FDR检验,然后突出显示p<=0.05的,q<=0.05的
补充,方法二,运行师姐的脚本进行FDR检验,运行前需要先做那个假基因的鉴定,运行非常快,结束后会生成三个结果文件。
python3 ~/Qxy/qxyjiaoben/fromzyr/pseudogene_count.py ~/Qxy/knowngenome/gongxianxing/0926bidui
#这个假基因鉴定之前运行过了的话可以忽略,之前没运行过的要运行后才能假基因鉴定
python3 paml_fdr.py Umbilicaria_muehlenbergii_genomic.fna /ifs1/User/wuqi/Qxy/knowngenome/gongxianxing/0926bidui Umbilicaria_antarctica_genomic.fna new_Umbilicaria_muehlenbergii.gff3 paml_siteresult.txt suffix(自己随便写的后缀)
#这个脚本是得到paml结果之后进行fdr校正,需要先挑出假基因,然后再去分析
python3 /ifs1/User/wuqi/Qxy/qxyjiaoben/fromzyr/paml_fdr.py Umbilicaria_muehlenbergii_genomic.fna /ifs1/User/wuqi/Qxy/knowngenome/gongxianxing/0926bidui Umbilicaria_antarctica_genomic.fna ../new_Umbilicaria_muehlenbergii.gff3 paml_siteresult_m7m8_all.txt site_all2063_m7m8三、快速进化基因分析
1)根据paml_result.txt可以筛选出w=999的基因名字,存为w999.txt列表
2)将相关基因转移至新目录下
mkdir w999/
dos2unix w999.txt #Windows系统下编写的txt会存在一些字符,linux识别不了,要转换一下
cd /ifs1/User/wuqi/Qxy/knowngenome/gongxianxing/0926bidui #这是工作目录
while read line; do cp gapfiltered0926/"$line" /w999/; done < w999.txt
注意!查看数目是否一致,经常w999.txt列表中最后一个老被漏掉,不知道为啥!
mv w999/ gapfiltered/ #套一个gapfiltered的假名字,因为师姐代码里写的,只能识别该目录下的序列3)生成序列文件
nohup python3 /ifs1/User/wuqi/Qxy/qxyjiaoben/fromzyr/paml_inputfile.py Umbilicaria_muehlenbergii_genomic.fna /ifs1/User/wuqi/Qxy/knowngenome/gongxianxing/0926bidui Umbilicaria_antarctica_genomic /ifs1/User/wuqi/Qxy/knowngenome/gongxianxing/0926bidui/1019RAxML_bipartitions.result branchsite &
#使用该脚本批量生成输入文件,主要是序列文件和树文件的调整4)生成控制文件
还是要看一下ctl文件的,重点看俩输入文件、输出文件、model模型设置,零假设就是0,备择假设是2,Nsite也要相应改动,其他不变,fix_omega为0
cd PAML_branchsite #进入该目录
for dir in */; do mkdir "$dir/branchsite_fix"; done #在该目录的每一个子目录下生成一个branchsite_fix文件夹
for dir in */; do mkdir "$dir/branchsite_nofix"; done #在该目录的每一个子目录下生成一个branchsite_nofix文件夹
在工作目录下,把控制文件ctl_branchsite_nofix和ctl_branchsite_fix处理好
#注意里面的参数设置
cd PAML_branchsite #进入该目录
find . -type d -name "branchsite_nofix" -exec cp ../ctl_branchsite_nofix {} \; #批量把ctl_branchsite_nofix放入branchsite_nofix目录下
find . -type d -name "branchsite_fix" -exec cp ../ctl_branchsite_fix {} \; #批量把ctl_branchsite_fix放入branchsite_fix目录下5)运行
nohup python3 /ifs1/User/wuqi/Qxy/qxyjiaoben/fromzyr/paml_run.py /ifs1/User/wuqi/Qxy/knowngenome/gongxianxing/0926bidui branchsite 20 & #运行paml6)卡方检验
原理是零假设结果文件branchsite_fix.mlc和备择假设结果文件branchsite_nofix.mlc中会有lnL(ntime: 47 np: 50): -4040.875649 +0.000000这样一行lnL信息
lnL1-lnL0 满足卡方分布Chi^2 自由度df=np1-np0=1,计算p值 <=0.05则显著
脚本都在/ifs1/User/wuqi/Qxy/qxyjiaoben/analysis_pvalue下
1.运行summary_fix.py、summary_nofix.py、summary_fixw.py、summary_nofixw.py脚本
得到resultfix.txt、resultnofix.txt、result_fixw.txt、result_nofixw.txt结果文件
2.传输到本地,提取合并成一个结果文件,并计算得到pvalue、qvalue、零假设w、备择假设w、FDR等,筛选得到快速进化基因(p<=0.05且目标分枝w高于背景分枝w)(注:这里需要用EXCEL做一下分列、合并等)
3.批量计算p值时,利用pchisq(lnL1-lnL0,df,lower.tail=FALSE)公式,用R去计算
例:
lnL(ntime: 47 np: 49): -4042.185659 +0.000000 零 omega (dN/dS) = 0.53728
lnL(ntime: 47 np: 50): -4040.875649 +0.000000 备择 w (dN/dS) for branches: 0.54760 0.22335
pchisq(-4040.875649+4042.185659,1,lower.tail=FALSE)注意更改一下!!!这里卡方检验计算2倍的lnl差值
即pchisq(2lnl ,df,lower.tail=FALSE)同chi2 df 2lnl,其中2lnl=2*ln1-ln0的绝对值,site model的df是2,branchsite model的df是1,快速进化df也是1
(注:就是自己用两个lnl值作差,然后生成上面的这行代码)
4.批量生成p.R脚本,加权限,然后Rscript p.R > r.result.txt 运行
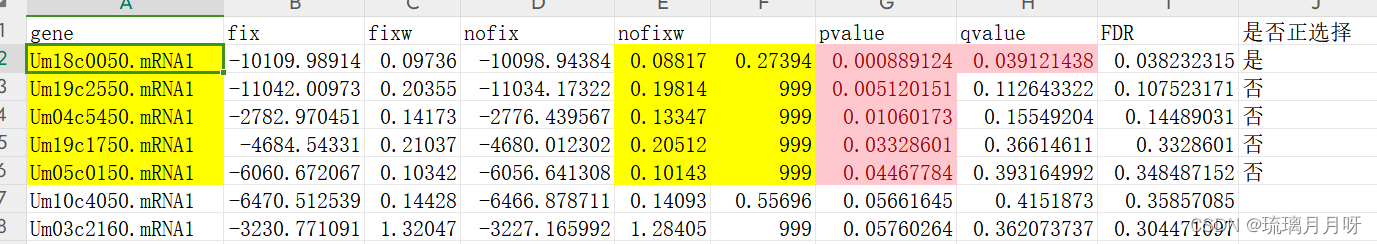 本地Excel结果整理
本地Excel结果整理
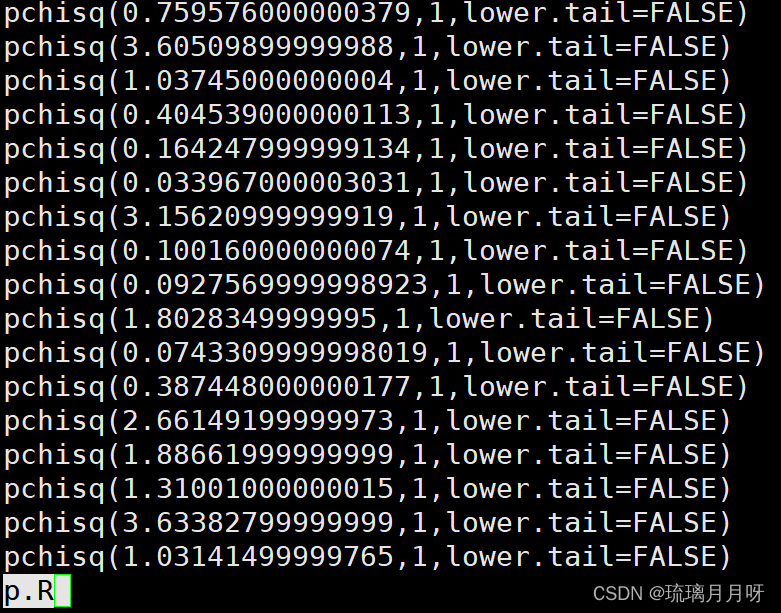 p.R批量计算pvalue脚本内容
p.R批量计算pvalue脚本内容
vim summary_fix.py
import os
result_file = 'resultfix.txt' # 汇总结果文件名
paml_dir = 'PAML_branchsite' # PAML_branchsite目录路径
# 遍历PAML_branchsite目录下的子文件夹
for subdir in os.listdir(paml_dir):
subdir_path = os.path.join(paml_dir, subdir)
if not os.path.isdir(subdir_path): # 如果不是文件夹,则跳过
continue
branchsite_fix_dir = os.path.join(subdir_path, 'branchsite_fix')
mlc_file = os.path.join(branchsite_fix_dir, 'branchsite_fix.mlc')
if not os.path.exists(mlc_file): # 如果branchsite_fix.mlc文件不存在,则跳过
continue
with open(mlc_file, 'r') as file:
lines = file.readlines()
lnL_line = None
for line in lines:
if 'lnL(ntime:' in line:
lnL_line = line.strip()
break
if lnL_line is not None:
with open(result_file, 'a') as result:
result.write(f'{subdir}\t{lnL_line}\n')
print('结果已汇总到result.txt文件中。')summary_fix.py系列脚本基本内容,就结果文件、输入文件、查找的内容不一样,看仔细了!!
四、进行site model分析
用的还是共线性比对流程下来得到的gapfiltered文件夹,本次有2063个文件(如果gapflitered有啥别的名字一定要提前改过来,师姐脚本只支持查找gapfiltered文件夹进行操作,且文件夹下只允许基因序列,其他的会报错)
1)批量准备输入文件paml_inputfile.py
#python3 paml_inputfile.py <参考序列文件名> <工作目录路径> <前景物种名称>(与文件中名字相对应,多个物种逗号分隔) <进化树文件路径> <模型>(branchsite、site)选一个
nohup python3 /ifs1/User/wuqi/Qxy/qxyjiaoben/fromzyr/paml_inputfile.py Umbilicaria_muehlenbergii_genomic.fna /ifs1/User/wuqi/Qxy/knowngenome/gongxianxing/0926bidui none /ifs1/User/wuqi/Qxy/knowngenome/gongxianxing/0926bidui/1019RAxML_bipartitions.result site &注意,site model 没有前景枝背景枝之分,要写 none ,而且模型写的时候是 site ,不能写 site model
2)批量准备控制文件
#nohup python3 paml_ctl.py 工作目录 site none &
nohup python3 /ifs1/User/wuqi/Qxy/qxyjiaoben/fromzyr/paml_ctl.py /ifs1/User/wuqi/Qxy/knowngenome/gongxianxing/0926bidui site none &#site model 不分零假设和备择假设,根据Nsites不同分几个子模型,不同的子模型之间进行比较(笔记上有图形解读)
#site model 一个控制文件全部跑出
3) PAML脚本批量运行
nohup python3 /ifs1/User/wuqi/Qxy/qxyjiaoben/fromzyr/paml_run.py /ifs1/User/wuqi/Qxy/knowngenome/gongxianxing/0926bidui site 20 &4)运行后的卡方检验
nohup python3 /ifs1/User/wuqi/Qxy/qxyjiaoben/fromzyr/paml_sitechi2.py /ifs1/User/wuqi/Qxy/knowngenome/gongxianxing/0926bidui &
















 4014
4014

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









