







前言:之前跑了共线性比对,得到了单拷贝直系同源基因,获取了四倍碱基位点进行建树,接下来咱们就跑mcmctree
输入文件1)含校准点的有根树 2)密码子在3个位点的多序列比对结果 3)mcmctree配置文件
流程:
cd ~/Qxy/knowngenome/orthofinder #先建几个文件夹^-^
mkdir mcmctree
cd mcmctree
一、含校准点的有根树
之前Orthofinder结果会得到单拷贝直系同源基因,24个石耳树+外群有642个,然后构建ML树
在figtree中打开树,在Tree选项那里,选择root trees以外群为根,选择order nodes,选择transform branches这个格式选择equal
然后选择Export tree导出树为nemick格式XX.trees
然后去timetree网站http://www.timetree.org/上查找分子时间,选择石耳属分化时间47.2MYA,U.decussata和其他石耳的分化时间为29.9MYA
调整好树格式和内容后上传至服务器
#共线性比对结果同理~(总之就是要有基因序列,比对,建树)
自己去改好格式再上传啊!!!
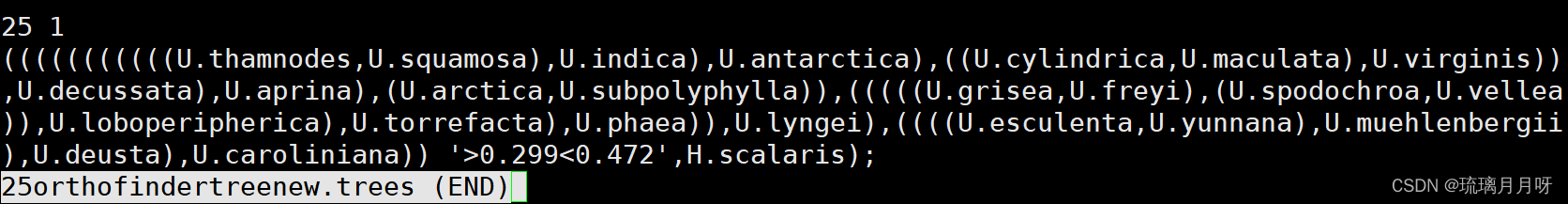
二、密码子在3个位点的多序列比对结果
根据看的教程和各种说明,理解是密码子在三个位点分别的比对结果,意思是氨基酸第一位列在一起,第二位列在一起,第三位列在一起,但是尝试操作后报错,说输入序列的phylip格式不对,因此还是用密码子去跑mcmctree
2.1用orthofinder比对结果去做:
2.1.1拿到25个物种的cds序列(拿到之后别的流程也可以直接用)
cd 进入对应目录,应该包含XX.cds-transcripts.fa文件
python3 ~/Qxy/qxyjiaoben/CDS2.py #提前改名字
sh ~/Qxy/qxyjiaoben/orthofinder_changename.sh #要提前更改一下名字
rm -rf modified_cds1.fasta #中间文件删掉
cp 一下转到同目录下#CDS2.py该脚本作用是把空格后面的那个删掉,从FUN_000001-T1 FUN_000001变成FUN_000001-T1(内容如下)
vim CDS2.py #脚本内容如下
from Bio import SeqIO
# 读取FASTA文件
records = SeqIO.parse('*.cds-transcripts.fa', 'fasta')
# 修改序列的描述符
new_records = []
for record in records:
seq_id = record.id
new_seq = str(record.seq)[:-11] # 去掉最后11个字符
new_record = f">{seq_id}\n{new_seq}"
new_records.append(new_record)
# 将修改后的记录写入新的FASTA文件
output_file = 'modified_cds1.fasta'
with open(output_file, 'w') as f:
f.write('\n'.join(new_records))#orthofinder_changename.sh 这是个老脚本了,之前Orthofinder的时候也用过(内容如下)
vim orthofinder_changename.sh
#!/bin/bash
# 设置物种名变量
species_name="Umbilicaria_antarctica" #这个应该是换哪个名就填哪个哈哈哈,也挺方便不是
# 读取蛋白文件中每一行文本
while read -r line; do
# 如果当前行包含 ">" 符号,说明是序列头部
if [[ "$line" == ">"* ]]; then
# 将序列 ID 和描述信息拆分出来,并在其前面加上物种名并连接
new_header=">${species_name}${line#>}"
echo "${new_header}" # 调试输出新的序列头部
else
# 否则说明是序列数据,直接输出即可
echo "${line}"
fi
done < "modified_cds1.fasta" > "Umbilicaria_antarctica_cds.fasta"<
 最低0.47元/天 解锁文章
最低0.47元/天 解锁文章


















 2102
2102

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









