







差分电荷密度、d带中心与COHP分析在材料设计中的应用
左七七
关键词:VASP;差分电荷密度;d带中心;COHP分析;材料设计
在催化科学、能源储存与转化、半导体器件和新能源材料研发领域,理解材料的电子结构是优化其性能的核心。差分电荷密度(Differential Charge Density)、d带中心理论(d-Band Center)与晶体轨道哈密顿布居(Crystal Orbital Hamiltonian Population, COHP)作为量子尺度的重要分析工具,可直观揭示化学键形成机制、电荷转移路径及催化活性位点特性,为材料理性设计提供理论基石。传统实验手段难以直接观测原子尺度电荷分布与轨道相互作用,而基于密度泛函理论(Density Functional Theory,DFT)的第一性原理计算成为破局关键。VASP(Vienna Ab initio Simulation Package)作为国际公认的电子结构计算权威软件,在以下场景中展现独特优势(功能还有很多,这里只做部分介绍)(可视化软件使用开源软件VESTA,界面如图1所示):
1. 差分电荷密度:通过计算吸附/解吸附过程的电荷的转移,可以直观看出材料分界面处的电荷的得失(如图2、图3、图4所示)。当然,如果需要定量的计算,我们也可以通过计算Bader电荷得出相应结论。以此评估材料之间微观的相互作用。
2. d带中心理论:关联过渡金属d电子态与吸附能强度,定量预测催化活性;
3. COHP分析:量化化学键强度与轨道贡献(例如预测氧还原反应中金属原子与反应中间体的键合强度,如图5、图6所示)。通过成键态和反键态贡献的对比,评估中间体的吸附强度,有效地指导实验研究。
图1 VESTA软件操作界面图。
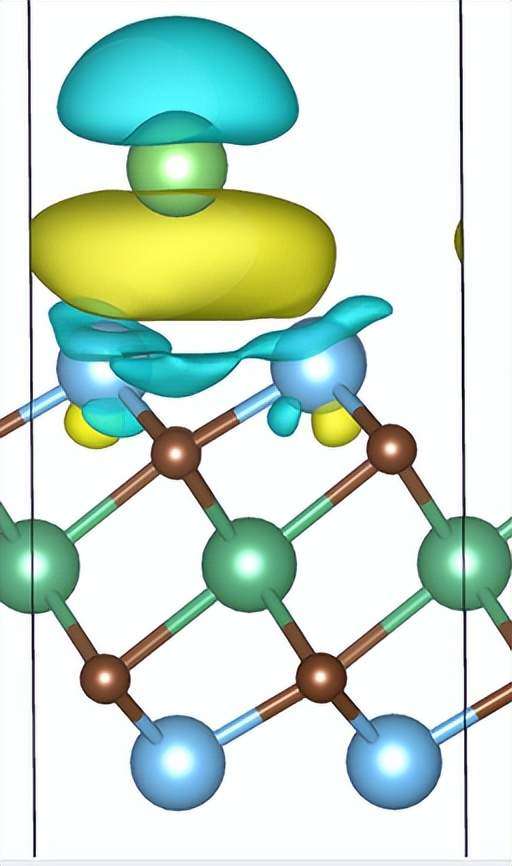
图 2 MXene结构上吸附Li原子的差分电荷密度图(这里只截取了结构的一部分)。
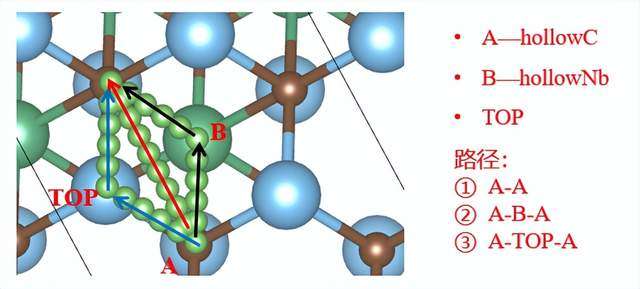
图3 金属离子在MXene上的扩散路径。
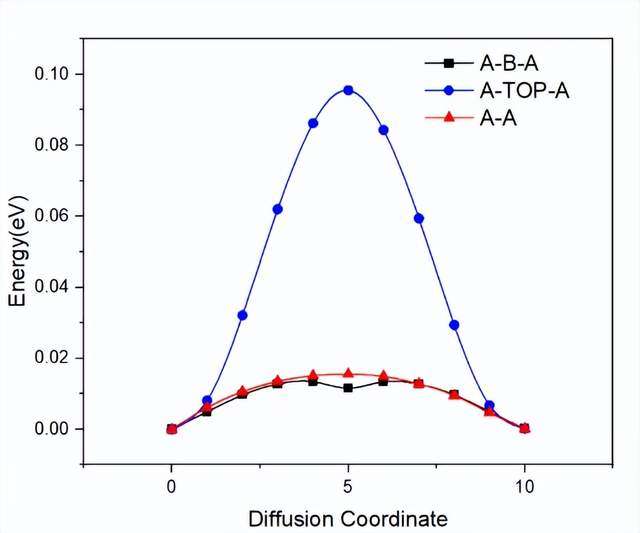
图4 金属离子在MXene上的扩散势垒。
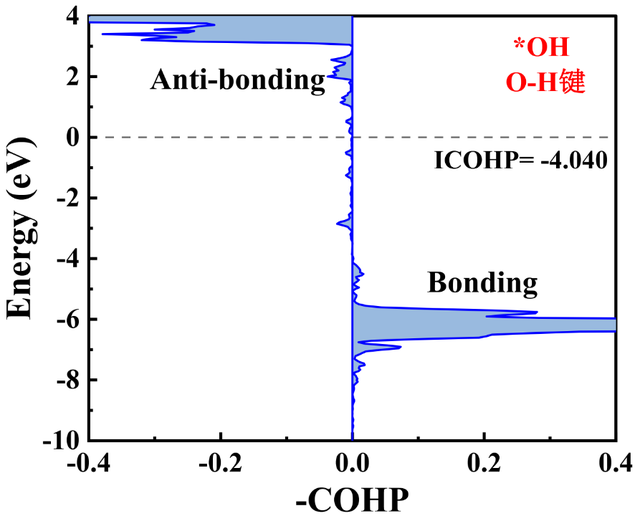
图5 活性位点Cr原子上吸附OH时O-H键的COHP分析。
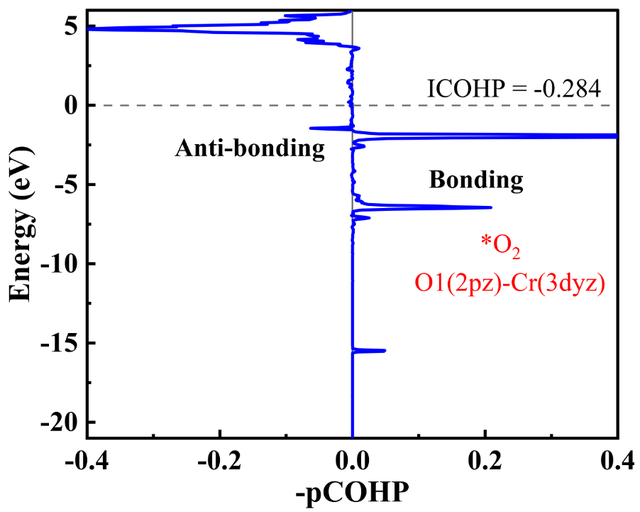
图6 活性位点Cr原子上吸附O2分子时Cr-O键的具体轨道的COHP分析。
我们的技术优势
与实验上相互配合:结合VASP电子结构计算,从微观角度分析活性机理,能够帮助解释实验中的相关结论;
定制化分析:针对催化、电池、光电等场景根据所给体系有专门的差分电荷与COHP解析流程;
数据结果可视化:提供出版级差分电荷等值面图、COHP分析图等。
服务对象
本案例适用于:
• 催化材料(单原子催化剂、合金/氧化物复合体系)
• 能源存储(锂离子电池电极/电解质界面、锂硫电池多硫化物锚定)
• 低维材料(二维异质结、拓扑绝缘体表面态)
• 光电转化(钙钛矿/有机半导体界面载流子动力学)
联系我们
若您需要:
• 解析材料性能背后的电子结构机制;
• 通过量子计算指导实验合成路径;
• 提升论文理论深度与创新性……
欢迎关注微信公众号“320科技工作室”,我们提供从建模、计算到数据可视化的全流程解决方案。


















 1337
1337

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











