









我用的系统是Ubuntu版本
安装软件 fftw-3.3.10 lammps-2Aug2023 mpich-4.2.0b1
centos版本apt命令换成yum
一、准备工作
1.下载需要的依赖包
sudo apt-get install gcc
sudo apt-get install g++
sudo apt-get install gfortran
sudo apt-get install make
2.下载需要的压缩包
FFTW下载地址:http://www.fftw.org/download.html
MPICH下载地址:https://www.mpich.org/downloads/
LAMMPS下载地址:https://www.lammps.org/download.html
3.将三个压缩包传到linux系统下的Downloads目录下,打开终端进入Downloads,使用下面命令解压,注意文件名
tar -zxvf fftw-3.3.10.tar.gz
tar -zxvf mpich-4.2.0b1.tar.gz
tar -zxvf lammps-2Aug2023 .tar.gz
二、安装fftw
进入Downloads目录下开始安装
cd fftw-3.3.10 #进入解压后的文件目录
./configure --prefix=/usr/local --enable-float #配置FFTW安装路径,这里是/usr/local
sudo make
sudo make install
三、安装MPICH
进入Downloads目录下开始安装
cd mpich-4.2.1 #进入解压后的文件目录
./configure --prefix=/usr/local #配置FFTW安装路径,这里是/usr/local
sudo make
sudo make install
四.安装lammps
1. 进入Downloads目录下开始安装
cd lammps-2Aug2023/src #进入解压后文件夹的src
make package-status
make yes-包名 #选择所需要的依赖包
2.打开src/MAKE目录下makefile.mpi文件
sudo vim Makefile.mpi
3.更改为以下内容

注意自己的安装路径

4返回目录src,使用安装命令
sudo make mpi #使用mpi并行文件
运行成功后自动生成lmp_mpi文件
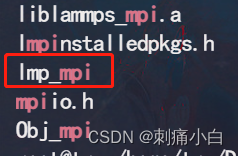
五,测试
我们到lammps自带的例子中试着跑一下
mpirun -np 4 lmp_mpi < in.crack
## lmp_mpi为自动生成文件的路径
## in.crack为examples/crack路径下的文件
成功运行截图
















 7128
7128

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









