







LAMMPS(Large-scale Atomic/Molecular Massively Parallel Simulator),即大规模原子分子并行模拟器,是一款由美国Sandia国家实验室开发的经典分子动力学代码。它主要用于模拟气体、液体和固体状态下粒子的集合行为,能够处理全原子、聚合物、生物、金属、粒状和粗粒化体系。
LAMMPS是目前用于分子动力学模拟的常用软件之一,网上安装教程五花八门,质量良莠不齐。本次教程旨在提供高效可用的安装部署方法,对初级使用Linux系统以及刚接触科学计算类软件用户十分友好。
如何获取LAMMPS
Lammps是开源软件,官方链接参考:https://www.lammps.org
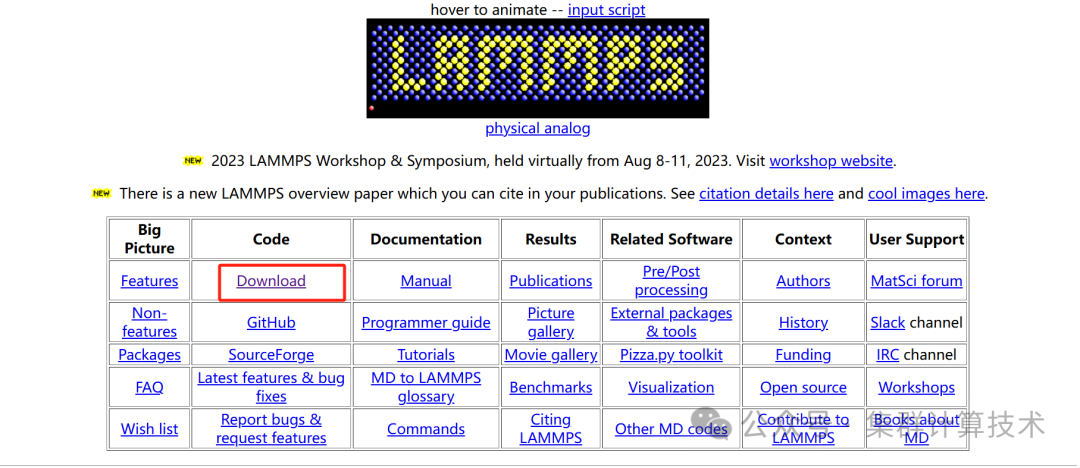
进入下载的页面,历史版本可以通过:
https://github.com/lammps/lammps/releases 获取对应的版本
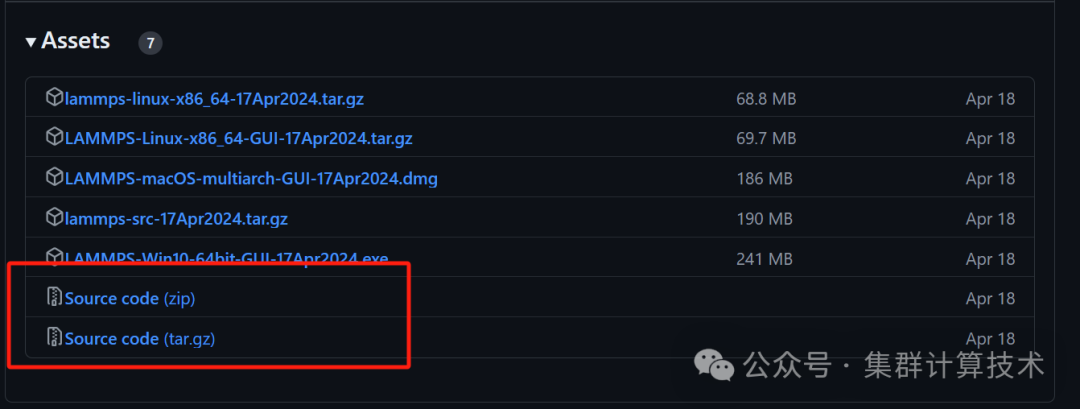
为了更加灵活的构建或者扩展LAMMPS,建议下载源码,从源代码级别重新构建。
安装编译环境 Intel oneAPI
详细可以参考
安装部署oneAPI
 最低0.47元/天 解锁文章
最低0.47元/天 解锁文章


















 4363
4363

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









