







文献介绍

文献题目: ezSingleCell: an integrated one-stop single-cell and spatial omics analysis platform for bench scientists
研究团队: 陈金妙(新加坡科学技术研究局)
发表时间: 2024-07-03
发表期刊: Nature Communications
影响因子: 14.7
DOI: 10.1038/s41467-024-48188-2
摘要
ezSingleCell 是一款交互式且易于使用的应用程序,用于分析各种单细胞和空间组学数据类型,无需具备编程知识。它结合了性能最佳的公开可用方法,用于深入的数据分析、整合和交互式数据可视化。ezSingleCell 由五个模块组成,每个模块都设计为针对一种数据类型或任务的综合工作流程。此外,ezSingleCell 允许统一接口内不同模块之间的串扰。可接受的输入数据可以采用多种格式,而输出则由可供发表的图形和表格组成。提供深入的手册和视频教程来指导用户分析工作流程和参数调整,以适应他们的研究目标。ezSingleCell 的简化界面可以在不到五分钟的时间内分析包含 3000 个细胞的标准 scRNA-seq 数据集。ezSingleCell 有两种形式:免安装的 Web 应用程序(https://immunesinglecell.org/ezsc/)或带有 shinyApp 界面的软件包(https://github.com/JinmiaoChenLab/ezSingleCell2)用于离线分析。
前言
scRNA-seq 已成为一种在单细胞水平获取基因表达谱的强大技术。这使得我们能够对生物系统的异质性有新的见解。单细胞分析也已扩展到其他组学,例如单细胞 ATAC-seq 和 CITE-seq,分别用于测量染色质可及性和蛋白质。此外,空间转录组学的进步现在使研究人员能够以单细胞分辨率探测组织样本,同时保留其空间背景。后一种能力对于理解不同细胞类型如何在空间上排列以产生组织的新兴特性至关重要。
单细胞技术的迅速崛起导致了大量数据的产生。与此同时,正在开发新的工具来分析生成的数据并产生新的生物学见解。2021 年,Zappia 和 Theis 报告称,scRNA 工具数据库中编目的单细胞工具数量超过 1000 个。目前,两个软件平台仍然占据主导地位,即 Seurat 和 Scanpy,它们分别是 R 和 Python 中单细胞分析的事实上的标准。两者都需要最低水平的生物信息学专业知识和编程知识,从而为基础科学家的数据分析带来了障碍。相比之下,具有直观图形用户界面的工具将极大地有利于希望进行单细胞实验的实验室科学家。研究界和商业公司最近开发了几种网络服务(Supplementary Table 1),但大多数仅限于转录组学分析,无法处理空间组学、单细胞多组学和单细胞染色质可及性( scATAC-seq)以统一的接口导出数据。此外,这些网站主要提供质量控制、数据聚类和降维等基本分析功能,而诸如细胞类型识别和细胞间相互作用等更高级的下游分析并不包含在其管道中。只有 ICARUS 和 Cellar 可以处理多种数据模式并提供一些下游分析,例如细胞类型识别,但不能提供其他分析,例如细胞间通信和空间数据集的细胞类型反卷积。另一个网络服务 SciAp (https://humancellatlas.usegalaxy.eu/ ) 集成了来自不同工作流程的工具,包括 Scanpy 的 20 个模块,涵盖数据过滤、归一化、可变基因选择、聚类、降维和轨迹推理方法,但是目前仅处理 scRNA-seq 数据。此外,当前的网络服务无法实现不同数据模式的整合分析,例如将 scRNA-seq 与空间转录组学相整合。
为了提供更全面的数据分析平台和用户友好的界面,作者推出了 ezSingleCell,这是一个为实验室科学家提供的集成的一站式单细胞和空间分析网络服务 (https://immunesinglecell.org/ezsc/)。 ezSingleCell 接受多种格式的数据输入,例如文本格式(csv 和 tsv)或 10x Cell Ranger/Space Ranger/Cell Ranger-ATAC 输出,并返回可发表的图形和表格。ezSingleCell 在以下方面对现有的单细胞数据分析 web 服务(包括 SciAp、ICARUS、CELLAR)进行了改进。首先,ezSingleCell 涵盖范围更广的单细胞数据分析,即单细胞多组学、单细胞 ATAC-seq 和空间转录组学。用于这些分析的工具包括作者团队的内部算法、GraphST 和 CELLiD,以及通过基准研究确定的性能最佳的公开工具,例如 Seurat、Harmony、scVI、CellphoneDB、MOFA + 和 Signac。其次,ezSingleCell 提供了许多超出基本分析流程的高级分析功能。所有模块共有的分析选项包括差异基因表达分析、基因集富集、细胞类型相似性和细胞间通讯。此外,ezSingleCell 还提供特定于模块的分析,例如 scATAC-seq 模块中的 Peak2GeneLinkage 以及空间数据集的细胞类型反卷积。第三,scATAC-seq 数据集可以使用 ezSingleCell 进行分析,这是大多数网络服务缺乏的功能。第四,ezSingleCell 可以使用 geometric sketching 扩展到大型数据集。Geometric sketching 对跨越一百万个或更多细胞的大型 scRNA-seq 数据集进行下采样,同时保留稀有的细胞状态。该技术对于加速大型数据集的聚类、可视化和整合分析非常有用。人们还观察到,geometric sketching 在通过聚类区分生物细胞类型方面始终有效。最后,ezSingleCell 允许不同分析模块之间的串扰。例如,处理和分析的带注释的单细胞 RNA-seq 数据可用于对空间数据中的细胞类型进行反卷积,或执行标签转移以执行 scATAC-seq 数据的细胞类型注释。目前,还没有网络服务能够在统一的界面中将两种组学数据类型链接在一起。
研究结果
1. ezSingleCell 概述及其相对于其他工具的优势
ezSingleCell 由五个模块组成,single-cell RNA-Seq (scRNA-seq)、single-cell data integration (scIntegration)、spatial transcriptomics (ST)、single-cell multiomics (scMultiomics)、single-cell ATAC-seq (scATAC-seq)(Fig. 1)。每个模块都提供了用于完整数据分析工作流程的多种工具,从数据预处理到交互式结果可视化。对于每个分析步骤,作者根据他们和其他基准研究选择了表现最好的方法。ezSingleCell 中可用的已发表和内部新颖算法列于 Supplementary Table 2。对于每个分析步骤,作者提供了适合大多数分析的默认参数,但用户也可以调整它们以获得最佳结果。在 scRNA-seq 分析模块中,用户可以执行基本分析例如聚类和差异基因表达分析,以及高级分析例如使用作者内部新颖算法 CELLiD 或 CellTypist 进行细胞类型识别、基因集富集分析 (GSEA)、细胞间通讯分析。作者还整合了 'clustree' 包来帮助用户选择与其生物学问题相关的最佳 clusters 数。scIntegration 模块提供单细胞转录组学中性能最高的四种数据整合算法,即 Seurat Integration、Harmony、scVI、fastMNN。作者还提供了 iLISI 等定量指标,供用户评估批量整合性能。空间转录组学 (ST) 模块配备了 Seurat 的空间转录组学分析功能和作者内部的 GraphST 算法。这些工具可以对使用不同技术平台(例如 10x Genomics Visium)和亚细胞技术(例如 10x Genomics Xenium)获取的空间数据进行空间聚类和细胞类型反卷积。对于 scMultiomics 模块,作者评估了所有可用的方法并选择了 Seurat WNN 和 MOFA + 进行包含。这些方法可以处理多模态数据,包括 CITE-seq(联合 scRNA-seq 与蛋白质)和 10x Multiome(联合 scRNA-seq 与 ATAC-seq)。最后,scATAC-seq 模块提供 Signac 的功能,用于单细胞染色质可及性数据的端到端分析,包括峰识别、定量、质量控制、降维、聚类、与单细胞基因表达数据集的整合,DNA motif 分析和交互式可视化。
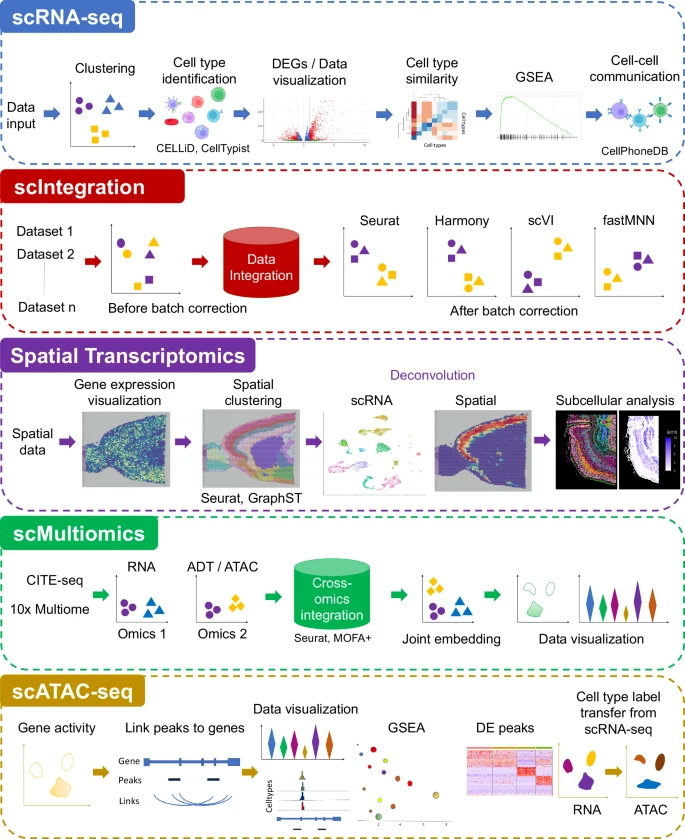
ezSingleCell 包含五个模块:single-cell RNA-seq (scRNA-seq)、single-cell data integration (scIntegration)、Spatial Transcriptomics、single-cell Multiomics (scMultiomics)、single-cell ATAC-seq (scATAC-seq)。该图还显示了每个模块可以执行的主要任务以及每个模块中可用的工具。
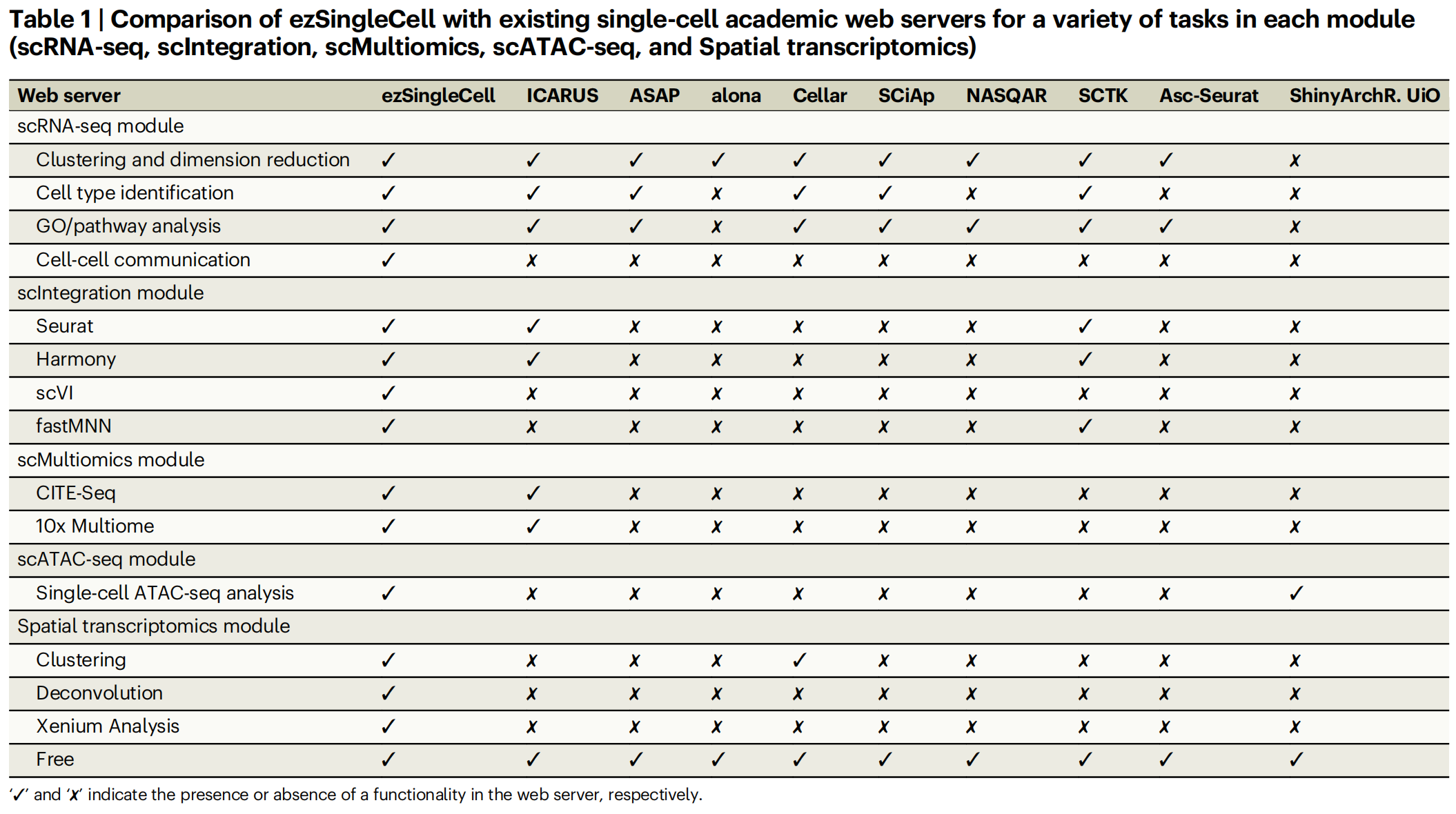
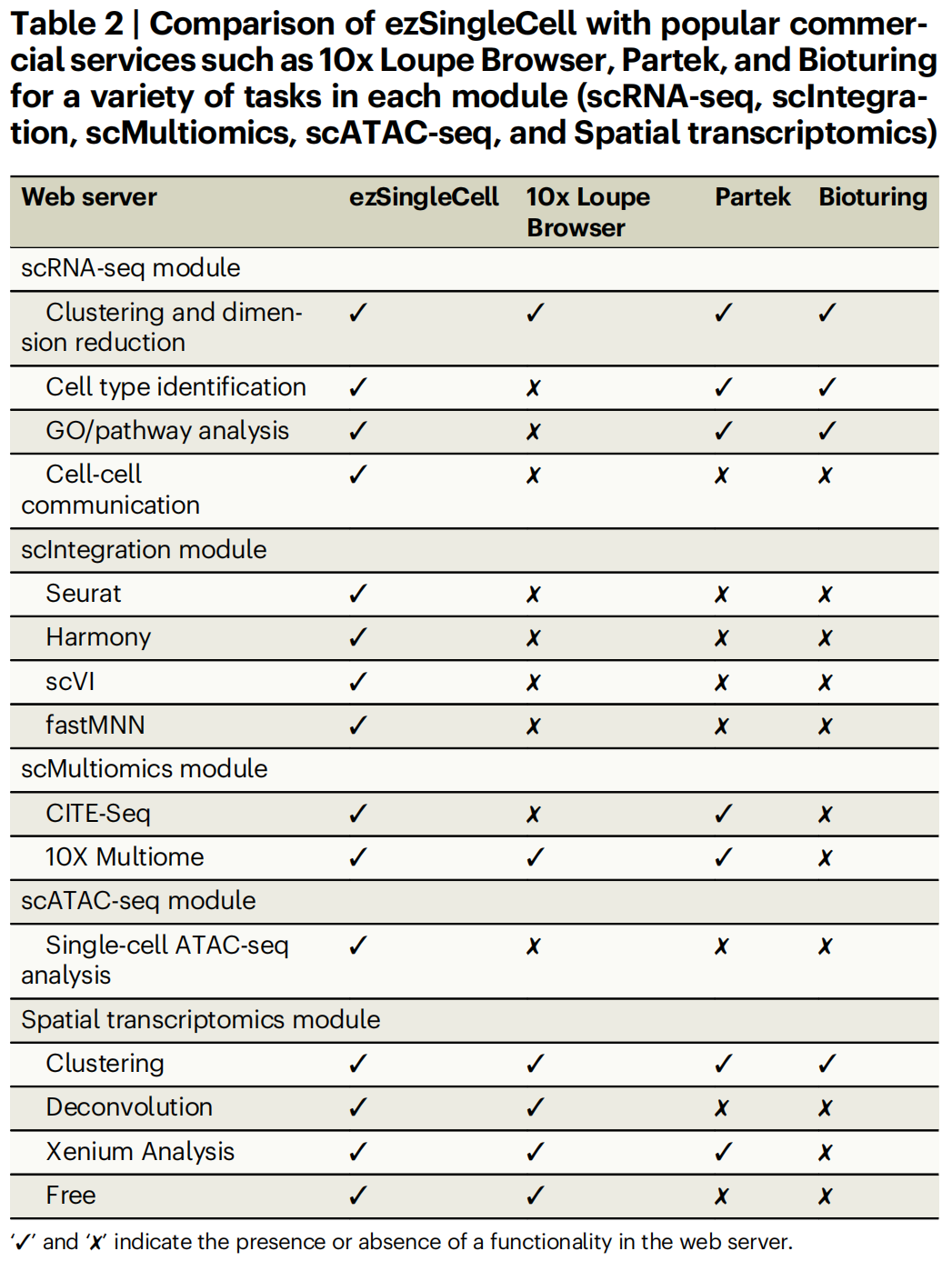
在这里,作者广泛地将 ezSingleCell 的功能与公开可用的网络服务和商业服务进行比较。如 Tables 1 and 2 所示,大多数 Web 服务(例如 SciAp、ASAP、alona、NASQAR、SCTK、Asc-Seurat)仅限于转录组学分析。一些 Web 服务确实提供了额外的功能,例如数据整合(SCTK 2.0、ICARUS)、sc-multiomics(ICARUS、Cellar)、scATAC-seq(shinyArchR.UiO)和空间分析(Cellar),但它们不提供用于综合分析的统一界面。现有的网络服务也不允许不同分析模块之间的相互作用,例如使用单细胞 RNA-seq 数据对空间转录组数据进行反卷积,或使用单细胞 RNA-seq 数据对 scATAC-seq 数据执行细胞类型标签 transfer。ezSingleCell 还在其模块内提供先进的下游分析功能。此外,ezSingleCell 还提供大多数网络服务所缺乏的 scATAC-seq 数据集分析支持。最后,ezSingleCell 的界面告诉用户哪些功能是可选的,哪些功能需要顺序运行,哪些可以并行运行。
ezSingleCell 有两种形式:免安装 Web 应用程序 (https://immunesinglecell.org/ezsc/) (Supplementary Figs. 1, 2) 和带有 Shiny 应用程序界面的软件包 (https://github.com/JinmiaoChenLab/ezSingleCell2),可以在计算机上运行进行离线分析。ezSingleCell 的源代码也可以在 Zenodo 上找到 (https://doi.org/10.5281/zenodo.10785313)。
2. ezSingleCell 的 scRNA-seq 模块简化了 scRNA-seq 数据的分析
ezSingleCell 的 scRNA-seq 分析模块依赖 Seurat 进行基本分析,并依赖其他软件包/内部算法进行高级分析,例如细胞类型注释、基因集富集分析 (GSEA) 和细胞间通讯(Fig. 2A)。除了常用的功能外,ezSingleCell 还为用户提供其他功能,例如细胞周期评分和回归,以减轻细胞周期异质性的影响。在这里,作者说明了 ezSingleCell 在 scRNA-seq 分析中的实用性(Figs. 2, 3)。作为示例,作者使用了来自 Seurat 聚类教程的 2700 个外周血单核细胞 (PBMCs) 数据集(Supplementary Table 3; Supplementary Dataset 1)。ezSingleCell 允许用户使用 min.genes 和 min.cells 等参数执行质量控制并过滤掉低质量细胞,并使用小提琴图、特征图和山脊线图可视化数据(Supplementary Fig. 3A)。然后,用户可以使用 log-normalization 或 SCTransform 执行数据预处理,并选择所需数量的高变特征。在此示例中,作者使用 min.genes 的默认参数值为 200,min.cells 的默认参数值为 3。然后,使用 log-normalization、前 2000 个高变基因的高变特征选择以及数据 scaling 来执行数据预处理。随后通过主成分分析 (PCA) 进行降维。然后使用前 10 个 PC 维度进行细胞聚类,k 最近邻值为 10,聚类分辨率为 2,以获得 15 个聚类。使用 PC 维度 1:10 生成的 UMAP 图对结果进行可视化(Fig. 2B)。
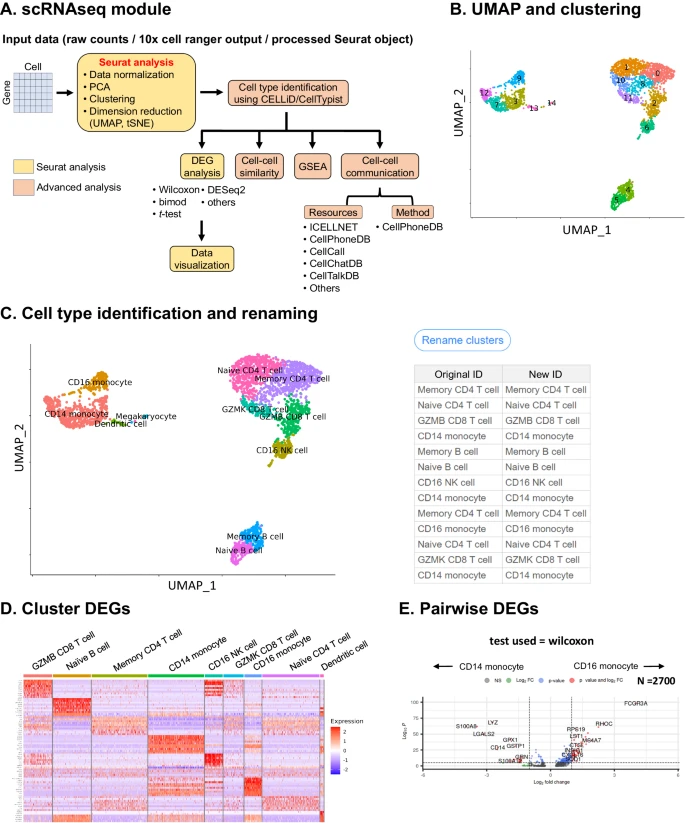
(A) scRNA-seq 分析的工作流程。
(B) ezSingleCell 中的 scRNA-seq UMAP 和聚类可视化。
(C) 使用 CELLiD 和 CellTypist 进行细胞类型识别。用户还可以在 ezSingleCell 中重命名 clusters。
(D) 使用 'wilcoxon' 检验进行聚类差异表达基因 (DEG) 分析。
(E) 使用 'wilcoxon' 检验对两种感兴趣的细胞类型进行配对 DEG 分析。
接下来,作者使用他们的内部细胞识别算法 CELLiD (https://www.immunesinglecell.org/cellpredictor), 使用 '血液' 细胞类型参考和 CellTypist (https://www.celltypist.org/) 来注释 clusters。CELLiD 分配了以下标签:Memory CD4 T、CD14 monocyte、Memory B、Naive CD4 T、Naive B、CD16 monocyte、GZMB CD8 T、CD16 NK、GZMK CD8 T、Dendritic cell、Megakaryocyte(Fig. 2C and Supplementary Fig. 3B)。使用 CellTypist,作者鉴定了 11 种细胞类型,即 MAIT cells、B cells、Tcm/naïve helper T cells、non-classical monocytes、CD16 NK cells、Tem/Trm cytotoxic T cells、Tem/Effector helper T cells、classical monocytes、Tcm/naïve cytotoxic T cells、DC、megakaryocytes/platelets。两种细胞类型注释方法都概括了 Seurat 教程中提供的原始注释。作者注意到,B cells 可以分为 naïve 和 memory B 子集,CD8 T cells 可以分为 GZMK 和 GZMB CD8 T cell 子集,从而提供更高分辨率的细胞类型标记。然后,作者通过检查标记基因表达来确认注释的正确性(Supplementary Fig. 3F, G)。ezSingleCell 还为用户提供了根据自己的喜好重命名 clusters 或合并不同 clusters 的实用程序。此外,用户可以根据用户定义的参数细分感兴趣的 cluster,这将有助于定义细胞类型子集。然后,作者计算了差异表达的基因,并发现了 B 细胞亚群的 CD79A、CD79B 和 MS4A1 等标记,以及 CD14 monocytes 的 S100A8、S100A9、LYZ(Fig. 2D)。使用小提琴图、特征图和山脊线图对这些基因进行可视化,证实了它们在相应细胞类型 clusters 中的较高表达(Supplementary Fig. 3C–E)。用户还可以计算两种指定细胞类型之间的 DEGs,并使用火山图可视化 DEGs,其中突出显示具有最高差异表达的最重要基因(Fig. 2E)。例如,作者观察到与 CD14 monocytes 相比,CD16 monocytes 中的 FCGR3A、S100A8、LYZ 差异上调。
然后,作者计算了细胞类型相似性(Fig. 3A),这显示出与 UMAP 图中观察到的 clusters 的一致性。最大 cluster 中的细胞类型(主要由 T 细胞和 NK 细胞组成)与同一 cluster 中的其他细胞类型显示出较高的相似性得分,而孤立的 clusters(例如 megakaryocyte)与所有其他细胞类型显示出较低的相似性得分(Fig. 2C)。当用户识别未知细胞群并希望估计其与已知细胞类型的相似性时,这种细胞类型相似性分析非常有用。
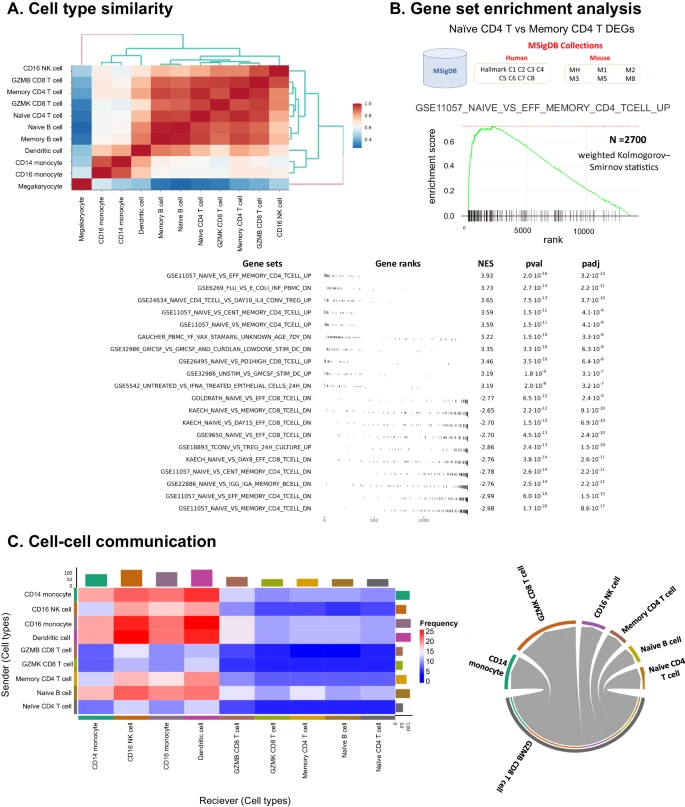
(A) 细胞类型相似性分析。
(B) 使用加权 Kolmogorov-Smirnov 统计进行基因集富集分析 (GSEA)。
(C) 使用 CellphoneDB 进行细胞间通信分析。
ezSingleCell 还提供 fgsea 用于基因集富集分析(Fig. 3B)。用户可以从 MSigDB 数据库中选择人类(Hallmark, C1-C8)和小鼠(MH, M1, M2, M3, M5, and M8)样本的任何基因集。在这里,作者使用人类 'C7' 免疫特征基因集将 GSEA 应用到 Naïve CD4 T 细胞和 Memory CD4 T 细胞之间的 DEGs。作者根据基因的对数倍数变化对基因进行排名,并计算最富集的途径。正如预期的那样,作者观察到与 Memory CD4 相比,GSE11057 NAÏVE VS MEMORY CD4 TCELL UP、GSE11057 NAIVE VS EFF MEMORY CD4 TCELL UP、GSE11057 NAIVE VS CENT MEMORY CD4 TCELL UP 基因集在 Naïve CD4 中的比例过高。Fig. 3B 列出了也显示出显着差异的其他基因集。最后,ezSingleCell 结合了广泛采用的 CellPhoneDB 包和各种配体-受体对数据库,用于分析细胞间通信。使用 CellPhoneDB,预测 cDC 和 CD16 单核细胞具有最高数量的潜在相互作用(Fig. 3C left)。然后,作者使用 p value cut-off <0.05 过滤 cDC 和 CD16 monocytes 之间的重要配体-受体对(Fig. 3C right)。
最后,scRNA-seq 模块的注释输出可以在其他模块中使用。例如,用户可以导航到 spatial transcriptomics 模块并使用单细胞注释来执行细胞类型/表型反卷积。
3. ezSingleCell 的 scIntegration 模块对多个 scRNA-seq 数据集进行批次校正
为了证明 ezSingleCell 中 scRNA-seq 数据的批次效应校正,作者使用了由 2 个批次组成的人类 PBMC 数据集。两个数据集均来自 10x Genomics,其中 3' 批次数据包含 8381 个细胞,5' 批次数据包含 7726 个细胞(Supplementary Table 3; Supplementary Dataset 1)。细胞类型注释是从以前的研究中检索的。对于批次效应校正,ezSingleCell 会同时接收表达数据集和带有批次信息的 metadata。细胞类型信息是可选的,但下游细胞类型分离评估是必需的。作者首先执行 log-normalization 的标准质量控制和数据预处理步骤,找到前 2000 个高度变化的基因,并使用默认参数进行 scaling。使用前 10 个 PCs 进行聚类,k 最近邻值为 10,Louvain 聚类分辨率为 0.6。在批次校正之前的 UMAP 图中,我们可以轻松观察到批次效应的存在(Fig. 4)。ezSingleCell 目前提供四种批次校正方法,即 Seurat、Harmony、scVI、fastMNN。作者运行了具有 2000 个 integration 特征的四种方法,并使用默认参数执行聚类和 UMAP 以可视化其结果。所有方法都能够成功消除批次效应(Fig. 4)。为了对批次混合进行基准测试,作者计算了 ezSingleCell 中 iLISI 分数的中位数。iLISI 指标测量局部区域内的批次数量,因此与批次数量匹配的分数表明混合良好。Harmony 排名第一 (iLISI = 1.70),其次是 fastMNN (1.54)、Seurat (1.43) 和 scVI (1.39)(Fig. 4)。批次整合后,可以进行其他下游分析,例如聚类、使用 CELLiD 算法进行细胞类型识别、差异基因表达分析和标记基因可视化(Supplementary Fig. 4)。
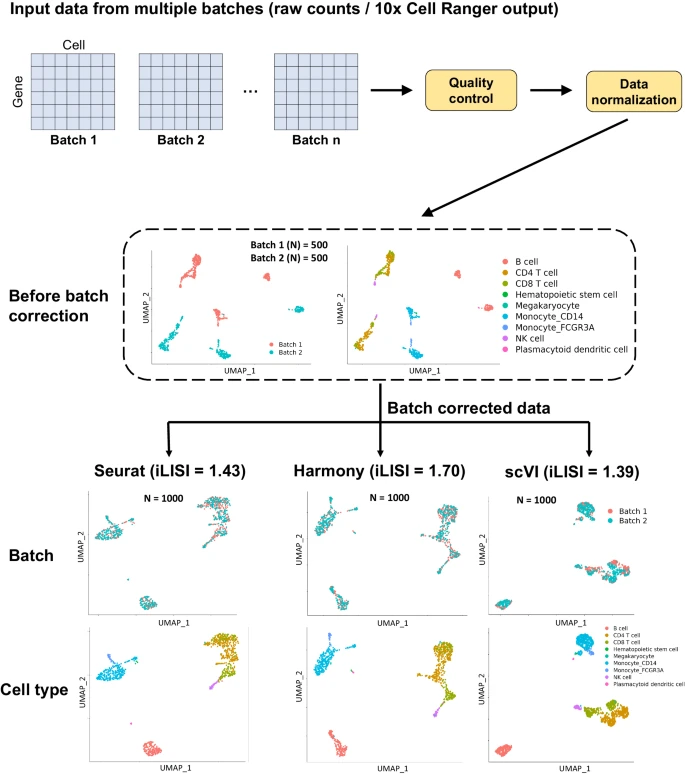
该模块的主要功能包括质量控制、标准化、批次效应校正前和批次校正后的 UMAP 可视化,使用 Seurat、Harmony、scVI,以及用于整合评估的 iLISI 评分。iLISI 分数越高表示批次混合和性能越好。
与 scRNA-seq 模块一样,scIntegration 模块的带注释输出可用于 spatial transcriptomics 模块中的细胞类型/表型反卷积。分析完成后,用户可以导航到 spatial transcriptomics 模块并使用单细胞输出。
4. ezSingleCell 的 ST 模块执行空间转录组学的聚类、整合和反卷积
在本模块中,作者结合了 Seurat 和 GraphST 来处理和分析空间转录组数据。Seurat 提供数据预处理和聚类的关键功能,而 GraphST 则执行空间信息空间聚类、多样本整合和细胞类型反卷积。GraphST 是一种最先进的图自监督对比学习方法,在与竞争方法的基准测试中取得了最佳性能。ST 模块可用于分析从不同平台获取的数据,例如 10x Genomics 的 Visium 和 Xenium,后者提供亚细胞分辨率。与其他模块类似,用户可以使用各种统计测试来计算聚类 DEGs(Supplementary Fig. 5D)和成对比较 DEGs(Supplementary Fig. 5E),并使用 'fgsea' 包对空间数据进行基因集富集分析(Supplementary Fig. 5F)。
在本演示中,作者分析了使用 10x Genomics Visium 采集并使用 Space Ranger pipeline v1.1.0 处理的小鼠大脑矢状前部数据集(Supplementary Table 3; Supplementary Dataset 1)。作者首先进行质量控制,然后使用 SCTransform 进行标准化,选择前 2000 个高度变化的基因,并进行数据 scaling。用户可以首先检查组织载玻片上的基因表达值(Fig. 5A)。接下来,作者使用默认参数执行 PCA。对于空间聚类,作者应用了 Seurat 和 GraphST。作者使用前 10 个 PC,k 最近邻值为 10,Louvain 聚类分辨率为 0.6,然后使用 UMAP 进行降维,以可视化找到的 15 个 clusters(Fig. 5B)。作者还将 GraphST 的聚类数量设置为 15,并观察到 GraphST 的聚类与手动注释更加一致(Fig. 5B)。
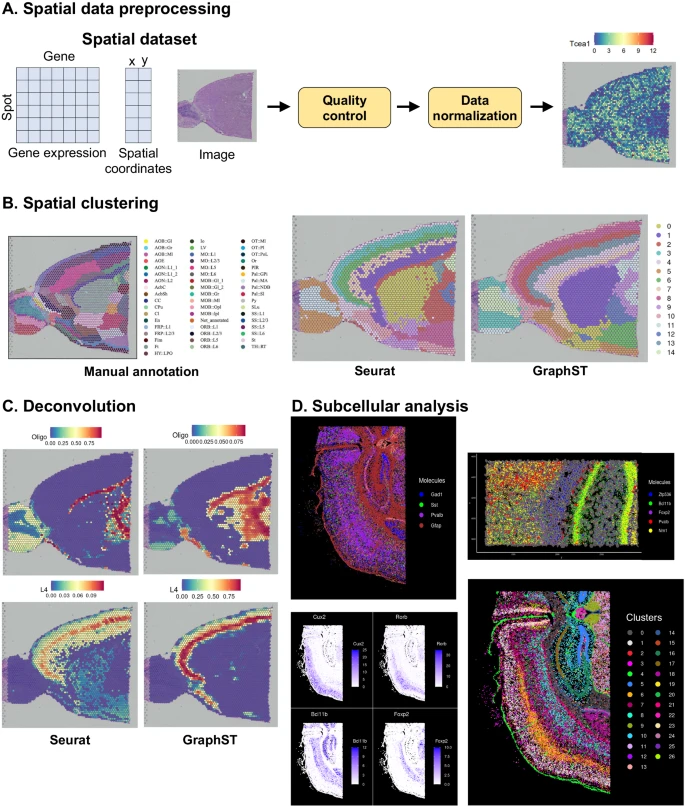
(A) 数据输入和预处理。
(B) 使用 Seurat 和 GraphST 进行空间聚类,并与病理学家手动细胞类型注释进行比较。
(C) 使用 Seurat 和 GraphST 进行细胞类型反卷积,显示使用 scRNA-seq 参考数据进行反卷积的细胞类型的比例。
(D) 亚细胞数据 (Xenium) 分析显示分子的聚类、分子表达谱的可视化以及细胞分割边界和单个分子的放大视图。
ezSingleCell 还提供使用带注释的单细胞参考对空间点中的细胞类型进行反卷积或注释的功能。为此,ezSingleCell 提供模块间可操作性,用户可以导航到 single-cell 模块,加载 scRNA-seq 数据集,执行数据处理和注释,然后返回 ST 模块进行细胞类型/表型反卷积。用户可以使用 Seurat 中的 'label transfer' 函数或 GraphST 中的 'project_cell_to_spot' 函数来执行反卷积,并且结果可以交互式可视化(Fig. 5C)。
ezSingleCell 支持对亚细胞分辨率数据(例如来自 Xenium 平台的数据)的分析。用户可以执行聚类分析并交互式地可视化亚细胞水平的表达模式。此外,用户可以放大以检查细胞组成和潜在的细胞间相互作用。用户还可以查看组织切片上每个基因的表达谱(Fig. 5D)。
5. ezSingleCell 的 scMultiomics 模块执行多种模态的联合分析
接下来,作者将演示 ezSingleCell 使用健康的 PBMC 数据集进行多模态单细胞分析。数据集包含基因表达和蛋白质表达 (CITE-seq) 数据,以及基因表达和染色质可及性 (MultiOme) 数据。两个数据集均从 10x Genomics 下载(Supplementary Table 3; Supplementary Dataset 1),其中 CITE-seq 数据集包含 7865 个细胞,MultiOme 数据集包含 3012 个细胞。CITE-seq 数据集使用带有默认参数的标准 log normalization 和带有 SCTransform normalization 的第二个 MultiOme 进行预处理。然后使用前 10 个 PC 维度、k 最近邻值 10 和聚类分辨率 0.6 执行细胞聚类(Fig. 6)。对于 CITE-seq 数据的联合模态分析,使用 Seurat WNN(RNA 和蛋白质相似性的加权组合)来计算用于聚类的 KNN 图。分辨率为 1.0 的聚类产生了 20 个 clusters,用于单独模态和联合分析。通过 CELLiD,作者鉴定了 16 种细胞类型,即 CD14 monocyte, CD16 monocyte, CD16 NK cell, Cycling T/NK cell, Dendritic cell, GZMB CD8 T cell, GZMK CD8 T cell, MAIT cell, Memory CD4 T cell, Naïve B cell, Naïve CD4 T cell, Naïve CD8 T cell, pDC, Plasma cell, Treg cell(Fig. 6)。接下来,作者对 B cells RNA 测定中的 MS4A1 和 ADT 测定中的 CD4 等相关标记进行可视化,以验证细胞类型注释(Fig. 6)。作者还使用 DEG 分析通过检查每个 cluster 的最高差异表达基因来进一步验证细胞类型。
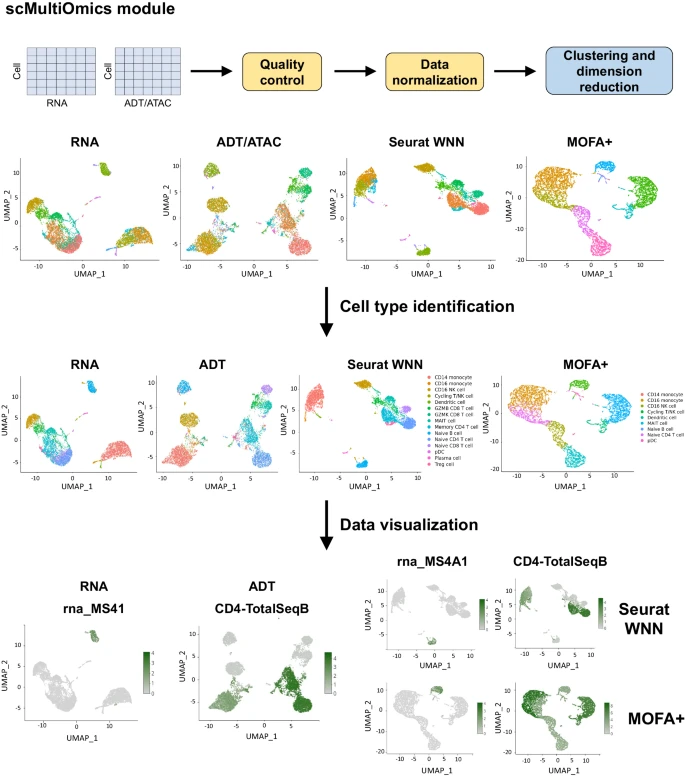
工作流程包括数据质量控制、预处理、聚类、降维、跨组学整合、整合后分析和可视化。目前,Seurat 加权最近邻 (WNN) 和 MOFA+ 可用于跨组学整合。整合后,可以使用 RNA 模态和Seurat WNN 或 MOFA + 的联合聚类来识别细胞类型。用户可以在联合 UMAP 中可视化特定基因和蛋白质。在这里,作者在 Seurat WNN 和 MOFA+ 的联合 UMAP 可视化中可视化了 B 细胞标记基因 MS4A1 和 CD4 T 细胞蛋白标记 CD4 的表达水平。
作者使用 MOFA+ 重新进行相同的分析,并通过将聚类数量(k 参数)指定为 10 并使用 MOFA + 中的所有潜在因子对数据进行聚类。使用 CELLiD,作者鉴定了 10 种细胞类型,即 CD14 monocyte, CD16 NK cell, Cycling T/NK cell, Dendritic cell, MAIT cell, Megakaryocyte, Memory CD4 T cell, Naïve B cell, Naïve CD4 T cell, pDC。作者再次在 RNA 和 ADT 测定中可视化标记基因,例如 B 细胞的 MS4A1 和 CD4 T 细胞的 CD4,以验证细胞类型注释(Fig. 6)。
6. ezSingleCell 的 scATAC-Seq 模块执行 scATAC-seq 数据分析和整合
在最后的演示中,作者使用 ezSingleCell 处理来自健康捐赠者的 10k PBMCs 的 scATAC-seq 数据集(Fig. 7A)。Cell Ranger ATAC 的输出是从 10x Genomics 下载的(Supplementary Table 3; Supplementary Dataset 1)。为了进行质量控制,用户可以计算核小体带型、转录起始位点 (TSS) 富集分数、峰中片段总数以及基因组 blacklist 区域中的 reads 比率等指标(Fig. 7B)。作者首先使用 Term Frequency-Inverse Document Frequency (TF-IDF) normalization 对数据进行标准化,然后进行特征选择和降维。然后使用前 10 个 PC 维度进行细胞聚类,k 最近邻值为 10,Louvain 聚类分辨率为 0.6。然后计算 UMAP 以可视化发现的 14 个 clusters(Fig. 7C)。接下来作者在 clusters 之间进行了差异峰值分析。然后使用小提琴图、特征图和覆盖图对结果进行可视化(Fig. 7D, E)。用户还可以使用各种统计测试来计算 cluster 之间的差异表达峰(DE peaks)(Fig. 7G)。
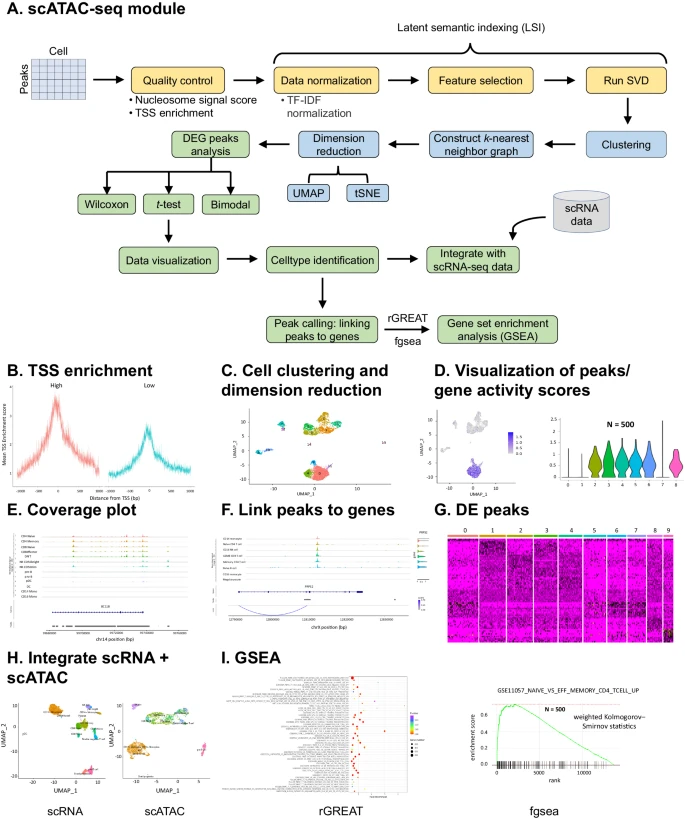
(A) scATAC-seq 分析的工作流程。
(B) 转录起始位点 (TSS) 富集。
(C) 数据聚类和降维。
(D) 数据可视化。
(E) 覆盖度图。
(F) 将峰与基因联系起来。
(G) clusters 间差异表达峰 (DE peak) 分析。
(H) 整合 scRNA-seq 和 scATAC-seq 数据以进行细胞类型识别。在这里,作者加载了处理后的人类 PBMCs scRNA-seq 数据集,并通过细胞类型标签 transfer 识别了 scATAC-seq 数据集中的 12 种细胞类型。
(I) 使用 rGREAT 和 fgsea 软件包对 scATAC-seq 数据进行基因集富集分析 (GSEA)。蜘蛛使用加权 Kolmogorov-Smirnov 统计进行 GSEA 分析。
ezSingleCell 还提供 scATAC-seq 和 scRNA-seq 模块之间的模块间可操作性。用户可以导航到 scRNA-seq 模块,加载并处理 scRNA-seq 数据集,然后返回到 scATAC-seq 模块以执行细胞类型标签 transfer 以进行细胞类型注释。在此示例中,作者加载了经过处理的人类 PBMCs 的 scRNA-seq 数据集,并用它来识别 scATAC-seq 数据中的 12 种细胞类型,即 CD4 Naïve cell, CD4 Memory cell, CD8 Naïve cell, CD8 effector cell, Double negative T cell, NK cell, pre-B cell, B cell progenitor, pDC, Dendritic cell, CD14+ Monocytes, and CD16+ Monocytes(Fig. 7H)。
用户可以将峰与基因联系起来,并可视化每个 cluster 中特定感兴趣基因的信号(Fig. 7F),并使用 rGREAT 或 fgsea 执行基因集富集分析(Fig. 7I)。在 ezSingleCell 中,rGREAT(基因组区域注释富集工具)支持人类和小鼠两个物种,具有多种基因集集合和不同的 TSS 注释,例如 txdb:hg19,TxDb.Hsapiens.UCSC.hg19.knownGene,RefSeq:hg19,GREAT:hg19,Gencode_v19。rGREAT 使用两种细胞类型之间差异表达的基因组区域(或峰)作为输入,并将生物功能与该区域相关联。例如,作者计算了 Naïve CD4 T 细胞和 Memory CD4 T 细胞之间的 DEGs,并使用 rGREAT 进行了基因集富集分析,观察到与 Memory CD4 T 细胞相比,GSE11057 NAÏVE CD4 VS PBMC CD4 TCELL UP 基因集在 Naïve CD4 T 细胞中上调。与 scRNA-seq 模块一样,scATAC-seq 模块还提供用于基因集富集分析的 fgsea 软件包,用户可以从 MSigDB 数据库中选择任何人类或小鼠基因集。例如,作者使用基因活性矩阵计算了 Naïve CD4 T 细胞和 Memory CD4 T 细胞之间的成对 DEGs,并使用 'fgsea' 函数和人类 'C7' 免疫特征基因集进行 GSEA 分析,以获得 Naïve CD4 与 Memory CD4 相比 over-represented 的通路。
7. ezSingleCell 允许不同模块相互作用
ezSingleCell 允许模块间操作,其中用户可以在一个模块中执行数据分析并使用在另一模块中获得的结果。为了演示这一实用性,作者分析了使用 10x Genomics Visium 获取并使用 Space Ranger pipeline v1.1.0 进行处理的人类乳腺癌数据集(Supplementary Table 3; Supplementary Dataset 1)。ezSingleCell 目前实现了四种类型的交互(Fig. 8)。在 scRNA-seq 模块中的细胞类型识别步骤中,用户可以单击 (1) the "Go to Spatial Deconvolution" button 或 (2) the "Annotate cell types for ATAC-data" button。第一个选项将引导他们进行空间转录组学模块的反卷积步骤。此处,带注释的 scRNA-seq 数据将用作参考,使用 Seurat 或 GraphST 对空间数据中的细胞类型比例进行反卷积。第二个选项将带用户进入 scATAC-seq 模块的细胞类型识别步骤,使用 Signac 将细胞类型标签 transfer 到 scATAC-seq 数据上。scIntegration 模块中的标记数据同样可以进行 transfer。此外,作者还启用了双向交互。在空间转录组学模块的反卷积步骤中,用户可以单击 "Load and process user reference dataset" 按钮,该按钮将用户带到 scRNA-seq 模块以上传、分析和注释其参考 scRNA-seq 数据。由此产生的注释数据将可用于空间转录组学模块中的细胞类型反卷积。同样,scATAC-seq 模块的细胞类型识别步骤中的 "Load and process user reference dataset" 按钮会导航到 scRNA-seq 模块以进行 scRNA-seq 数据处理和分析。然后将处理后的 scRNA-seq 数据提供给 scATAC-seq 模块以进行细胞标签 transfer。
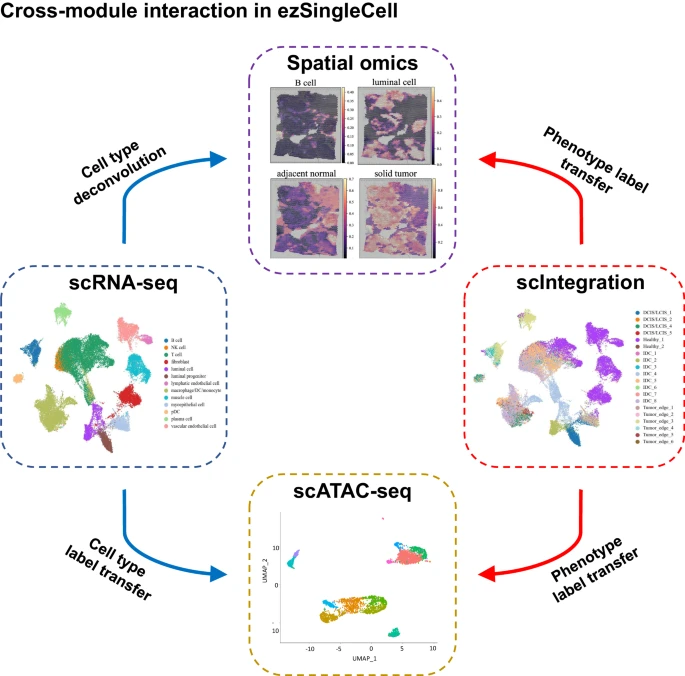
用户可以处理 scRNA-seq 数据,并使用单独的数据集或批次整合数据对空间组学数据中的细胞类型进行反卷积,或执行标签转移以完成 scATAC-seq 数据的细胞类型注释。
8. ezSingleCell 可以处理大型数据集而不影响性能
为了处理大型数据集,ezSingleCell 采用一种名为 'geometric sketching' 的技术对大型 scRNA-seq 数据集进行下采样,同时保留稀有细胞类型和细胞状态。通过在 ezSingleCell 中使用此功能,用户可以通过加速聚类、可视化和整合分析来执行大型数据集的数据分析。在 50,000 个细胞数据的用户测试场景中,用户可以在 5-6 分钟内完成包括聚类、降维、细胞类型识别在内的基础和高级单细胞数据分析,100,000 个细胞在 15 分钟内完成。
讨论
细胞的单细胞分析正在生成大量数据。尽管利用数据的分析工具越来越多,但基础科学家的数据分析仍受到这些工具的最低生物信息学技能要求的阻碍。ezSingleCell 是一个集成的一站式单细胞和空间组学分析平台,具有直观的 GUI,专为没有生物信息学背景的用户而设计。ezSingleCell 的分析模块涵盖不同单细胞组学实验生成的数据,即 scRNA-seq、scIntegration、scMultiomics(CITE-seq、10x Multiome)、scATAC-Seq 和空间组学。 ezSingleCell 通过结合内部新颖算法(例如用于细胞类型识别的 CELLiD 和用于空间聚类和反卷积的 GraphST)以及用于基本和高级分析的其他既定算法(例如批次效应校正、基因集富集分析、细胞通讯分析、空间反卷积)来实现这一目标。GUI 设计为用户友好型,可进行交互式数据探索和分析。用户可以使用各种参数自定义数据分析。ezSingleCell 还接受不同格式的输入,例如文本文件或 Cell Ranger / Space Ranger / Cell Ranger-ATAC 输出,并生成可供发表的图形和表格。ezSingleCell 有两种形式:免安装的 Web 应用程序(https://immunesinglecell.org/ezsc/),或带有 Shiny 应用程序界面的软件包(https://github.com/JinmiaoChenLab/ezSingleCell2),在计算机上运行以进行离线分析。ezSingleCell 的源代码也可以在 Zenodo 上找到 (https://doi.org/10.5281/zenodo.10785313)。
未来作者将继续对 ezSingleCell 进行维护和升级。随着每个实验分析的细胞数量迅速增加,这会导致生成非常大的数据集,因此对此类大数据的分析和集成非常耗时且占用大量内存。在 ezSingleCell 的未来版本中,作者将采用新颖的深度学习方法,以实现更高效的降维、聚类和批次整合。作者还将扩展当前的空间转录组学模块来分析空间蛋白质组学和空间多组学。作者还将添加基于空间邻近性和配体-受体对的表达来推断细胞间相互作用的功能。随着最新的亚细胞空间技术(例如 Nanostring CosMX、StereoSeq、Vizgen MERSCOPE、PixelSeq 和 SeqScope)的出现,作者计划采用新的算法来更好地进行细胞分割来处理此类数据集。
注:本文为个人学习笔记,仅供大家参考学习,不得用于任何商业目的。如有侵权,请联系作者删除。

本文由 mdnice 多平台发布
















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









