









R语言计算遗传距离
遗传距离是什么,怎样衡量
遗传距离指个体、群体或种之间用DNA序列或等位基因频率来估计的遗传差异大小。衡量遗传距离的指标包括用于数量性状分析的欧式距离(D),可用于质量性状和数量性状的Gower距离(DG)和Roger距离(RD),用于二元数据的改良Roger距离(GDMR)、Nei&Li距离(GDNL)、Jaccard距离(GDJ)和简单匹配距离(GDSM)等。
参考链接
遗传距离的计算
Roger遗传距离
先贴出计算代码:
library("poppr")
library("ape") # To visualize the tree using the "nj" function
library("magrittr")
library("adegenet")
library("pegas")
library("seqinr")
# data(microbov)
# caluclate Roger distance 短短三行代码
out<-read.vcfR("out.vcf")
dat <- vcfR2genind(out)
dfdist <- rogers.dist(dat)
我手上有hapmap文件,先转化成vcf文件,再进行计算,最后用as.matrix,write.csv读写出来,就得到了Roger遗传距离的矩阵。
解决过程
从未计算过遗传距离的我在网上找了很久,终于在一个问题下面找到了答案:
如何计算Roger遗传距离?使用poppr包来计算。

于是我继续搜索??poppr,得到了一系列接近答案的内容,在点进这个网站:http://grunwaldlab.github.io/Population_Genetics_in_R/Pop_Structure.html
我发现真的可以计算。
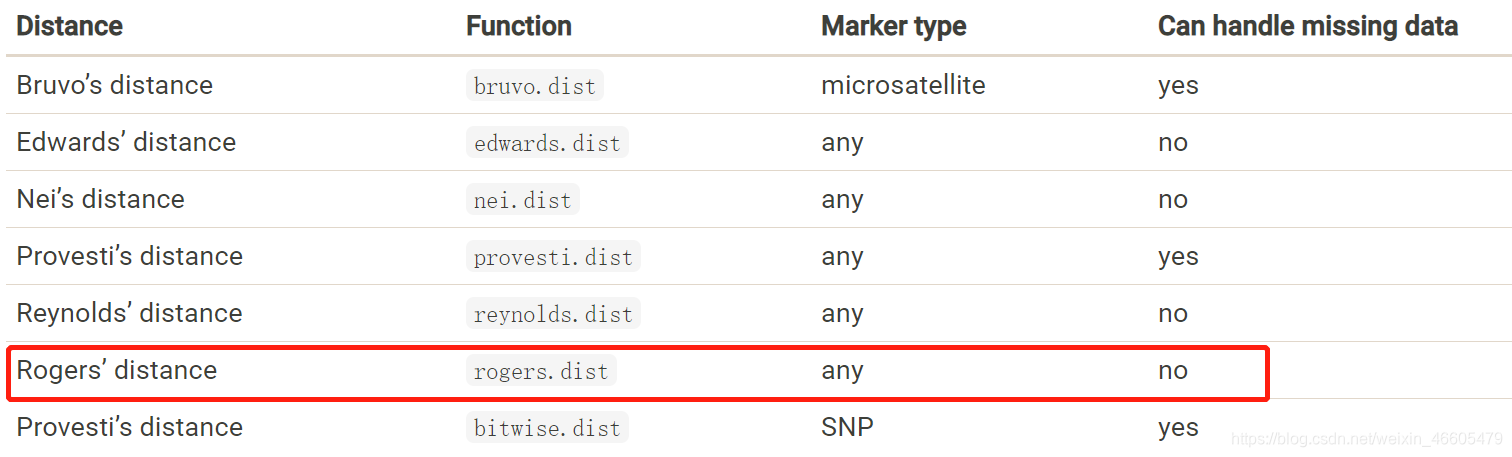
此外,还有不同计算遗传距离的方法:Bruvo’s distance、Edwards’ distance、Nei’s distance(也可以在MEGA里算)、Provesti’s distance、Reynolds’ distance、Provesti’s distance。打出来便于大家搜索。
参考链接:https://www.meiwen.com.cn/subject/nlsmwctx.html
关于一些弯路
一开始使用了data(microbov),发现是S4类型,但是用as.data.frame打开仅仅是基因型。所以一直想要把基因型直接转化为S4类型。于是刷到了这样一个帖子:
https://www.rdocumentation.org/packages/adegenet/versions/2.0.1/topics/df2genind
http://grunwaldlab.github.io/Population_Genetics_in_R/Pop_Structure.html

代码如下:
> temp <- lapply(1:30, function(i) sample(1:9, 4, replace=TRUE))
> temp <- sapply(temp, paste, collapse="")
> temp <- matrix(temp, nrow=10, dimnames=list(paste("ind",1:10), paste("loc",1:3)))
> temp
loc 1 loc 2 loc 3
ind 1 "1816" "9937" "5992"
ind 2 "7848" "1124" "2787"
ind 3 "3733" "8911" "5455"
ind 4 "6938" "1113" "3634"
ind 5 "5149" "4993" "1333"
ind 6 "8934" "5968" "9684"
ind 7 "5482" "3713" "2729"
ind 8 "1895" "7549" "4415"
ind 9 "4923" "5735" "3232"
ind 10 "6163" "2788" "6888"
> obj <- df2genind(temp, ploidy=4, sep="")
> obj
/// GENIND OBJECT /
// 10 individuals; 3 loci; 27 alleles; size: 10.2 Kb
// Basic content
@tab: 10 x 27 matrix of allele counts
@loc.n.all: number of alleles per locus (range: 9-9)
@loc.fac: locus factor for the 27 columns of @tab
@all.names: list of allele names for each locus
@ploidy: ploidy of each individual (range: 4-4)
@type: codom
@call: df2genind(X = temp, sep = "", ploidy = 4)
// Optional content
- empty -
## simple example
df <- data.frame(locusA=c("11","11","12","32"),
locusB=c(NA,"34","55","15"),locusC=c("22","22","21","22"))
row.names(df) <- .genlab("genotype",4)
df
obj <- df2genind(df, ploidy=2, ncode=1)
obj
tab(obj)
## converting a genind as data.frame
genind2df(obj)
genind2df(obj, sep="/")
可以试试看。

















 467
467

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









