







plink由哈佛大学的Shaun Purcell开发的一个免费,开源的全基因组关联分析软件。
官网:PLINK 1.9
主要功能:
1.数据提取,合并、提取特定SNP、样本、基因组某段区域的基因型3.;plink格式文件如何提取位点、删除位点、提取样本、删除样本 - 小鲨鱼2018 - 博客园 (cnblogs.com)
2. 数据质控(计算样本杂合度和SNP位点杂合度、最小等位基因频率MAF)
3. 格式转换;
4. 遗传参数
4.1 计算最小等位基因频率
4.2 计算杂合度
4.3 计算LD、过滤R2
4.4 计算亲缘关系IBS,构建G矩阵;
4.5 计算近交系数 ( 统计样本ROH);
5. PCA
6. plink将关联分析结果里的SNP注释:参考PLINK学习笔记 - 知乎
常用文件格式:
1.文本格式的 PLINK 数据包含两个文件:前缀名称必须一致
•包含相关个体及其基因型的信息(*.ped);格式转化为主等位基因0, 杂合1, 次等位基因2
•包含遗传标记的信息(*.map)。
map文件没有行头, 包括四列: 染色体, SNP名称, SNP遗传位置(CM), SNP物理位置(bp)
第一列 Family ID
第二列 Individual ID
第三列 Paternal ID (0表示无)
第四列 Maternal ID((0表示无))
第五列 Sex (1=male; 2=female; 0=unknown)
第六列 Phenotype(0/-9 表示无)
注:自己构建map文件plink格式的ped和map文件及转化为012的方法 - 知乎
2. 二进制 PLINK 数据由三个文件组成:
•包含样本标识符(ID)和基因型(*.bed)的二进制文件;基因型信息
•样本信息(*.fam);每一行就是一个样本。
•遗传标记文件( *.bim); 每一行是一个变异,及其注释信息。
bed fam bin 为一组
- --bfile
- bed是一个二进制的文件,与fam,bin文件互相对应。基因型用0,1表示,一般打不开,具体如下

00 ref 纯合
11 alt 纯合
01 缺失
10 杂合
- bim文件
0 Chr00:49209 0 49209 C T
0 Chr00:49287 0 49287 G T
该文件是在map的结果上在添加两列SNP位点

- fam
A01 A01 0 0 0 -9
A02 A02 0 0 0 -9
第一列 Family ID
第二列 Individual ID
第三列 Paternal ID (0表示无)
第四列 Maternal ID((0表示无))
第五列 Sex (1=male; 2=female; 0=unknown)
第六列 Phenotype(0/-9 表示无)
文件具体内容参考网站:
https://www.jianshu.com/p/2bf82e596e45
GWAS | 全基因组关联分析 | PLINK | 实战 | 统计遗传学_普通网友的博客-CSDN博客
基本命令概括
功能之一: 基因型数据提取、合并
1. 提取特定SNP的基因型:# snpid.list 只含1列SNPid,无列名称
-
--extract :提取指定标记
-
--exclude:剔除指定标记
-
plink --file plink --extract snpid.list --recode --out extract_snpid --chr-set 30 plink --file plink --exclude snpid.list --recode --out exclude_snpid --chr-set 30
-
2. 提取特定样本: #samplelist.txt 含2列 FID 个体IID,无列名称
-
--keep:只保留指定样本
-
--remove:剔除指定样本
-
samplelist.txt 1 0 sample1 2 0 sample2 3 0 sample3 4 0 sample4 plink --file plink --keep samplelist.txt --recode --out keep_sample --chr-set 30 plink --file plink --remove samplename.txt --recode --out remove_sample --chr-set 30
3. 基因组某段区域位点:
-
--from-bp, --from-kb, --from-mb:开始的物理位置,单位可以是bp、kb或mb
-
--to-bp, --to-kb, --to-mb:终止的物理位置,单位可以是bp、kb或mb
-
-
一、提取某个染色体上的SNPs plink --file plink --chr 1 --from-bp 500 --to-bp 100000 --recode --out 500bp_100000bp_SNP --chr-set 30 二、提取一个范围内的SNPs (从一个rs到另一个rs,必须在同一条染色体上) plink --bfile mydata --from rs273744 --to rs89883 --recode --out 根据染色体上的位置提取 plink --bfile mydata --chr 2 --from-kb 5000 --to-kb 10000 --recode --out 三、提取某个SNP上下20Kb的SNPs plink --bfile mydata --snp rs652423 --window 20 --recode --out 四、同时提取多个范围的的SNPs及单个SNPs (SNP名称或者范围内不能有空格) plink --bfile mydata --snps rs273744-rs89883,rs12345-rs67890,rs999,rs222 五、提取指定的SNPs 5.1 把SNP名称整理成单列的snpslist.txt文件 e.g. rs123 rs12345 rs123456 plink --file data --extract mysnps.txt 5.2把染色体、范围、基因名整理成txt文件 e.g.【行名不要】 CHR BP1(起始位置) BP2 (终止位置) LABEL(基因名or随便写) 1 123 456 ABC 12 5465 3456 DFG plink --file data --extract myrange.txt --range 输出plink格式时,后面都需要加上 --make-bed --out result
https://www.jianshu.com/p/43e7ca72ea71
4. 数据合并:
-
--merge:Plink格式数据前缀
-
--bmerge:二进制格式数据前缀
功能之二: 质控
vcftools过滤GWAS | 全基因组关联分析 | PLINK | 实战 | 统计遗传学_普通网友的博客-CSDN博客
1. 标记质控
-
--chr :指定保留哪些染色体上的标记
-
--freq 对所有位点进行基因频率统计
-
-missing 输出样本缺失率在 .imiss文件, 位点缺失率在 .lmiss文件.
-
--geno:缺失率在给定比例以下的标记将被剔除(0.1=10%)
-
--maf:最小等位基因频率在该值以下的标记将被剔除(0.01=1%) 基因频率进行的筛选
-
--hwe:哈德温伯格平衡检验P值低于该值的标记将被剔除 ;基因型频率进行的筛选(植物一般不宜筛选)
-
plink --noweb --file snp --het --out 5 --allow-extra-chr
2. 样本质控
-
--mind:缺失率在给定比例以下的样本将被剔除(0.1=10%)
-
plink --allow-extra-chr --noweb -file test --geno 0.05 --maf 0.05 --hwe 0.0001 --make-bed --out test1 # --geno 0.1 某个SNP在样本中缺失大于10%,删除该SNP:--geno # --mind 0.1 某个在某个样本中,SNP缺失大于10%,删除该样本:--mind
plink.imiss:个体缺失位点的统计
第一列为家系ID,第二列为个体ID,第三列是否表型缺失,第四列是缺失的SNP个数,第五列总SNP个数,第六列缺失率
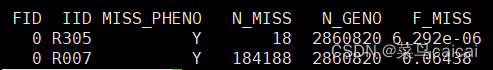
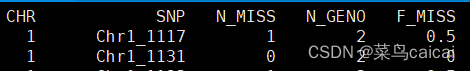
plink.lmiss:单个SNP缺失的个体数
第一列为染色体,第二列为SNP名称,第三列为缺失个数,第四列为总个数,第五列为缺失率。
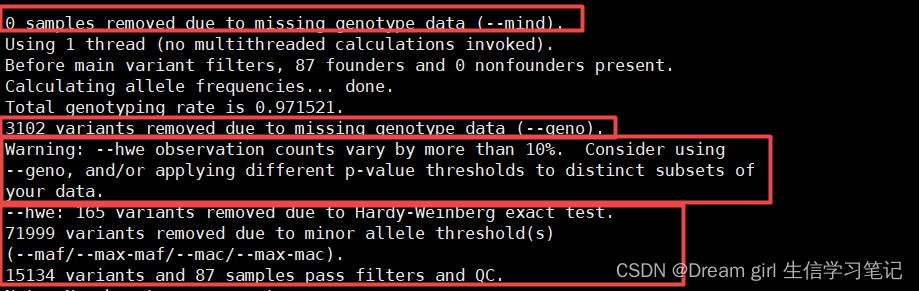
--hwe:
如果哈温质控的标记数超过最终剩余标记数的10%,就会有提醒
Warning: --hwe observation counts vary by more than 10%. Consider using --geno, and/or applying different p-value thresholds to distinct subsets of your data。
注意:所以质控的标准,过滤的标记都可以从日志文件里查看 .log
3.只保留SNP位点(剔除SNP以外突变类型,如indel)
-
--snps-only just-acgt:
-
只保留单位点突变的SNP,
-
加上just-acgt参数,则只保留等位基因型为A/T/C/G/a/t/c/g的位点
-
--chr-set:设置最大的常染色体数量(chr > 23)
-
--allow-extra-chr:允许不可识别的染色体编号,如鸡的Z和W染色体,出现除XY以外其它
参考网站
功能之三:格式转换
1.1 plink正常格式(map和ped)转二进制格式(b.bed, b.bim, b.fam)
比如这里有plink格式 a.map a.ped 将其转化为二进制文件:b.bed, b.bim, b.fam
plink --file a --out b- 如果染色体超过23,比如30对染色体,需要设定
--chr-set 30 - 如果有非数字染色体,比如性染色体,需要设定
--allow-extra-chr
1.2 plink二进制格式(b.bed, b.bim, b.fam)转为正常格式(map和ped)
plink --bfile b --recode --out c
- --bfile,因为输入文件b*为二进制,所以用--bfile,如果是一般格式,用--file即可
- --recode,要输出正常格式,所以用--recode指定,如果不加这个参数,默认是输出二进制文件
- --out,输出文件的前缀
1.3 正常plink文件(map和ped)转为vcf文件
plink --file c --recode vcf --out d
- --file,用--file指定正常plink格式的文件
- --recode vcf,要输出vcf文件格式
- --out,输出文件的前缀
1.4 二进制plink文件(b.bed, b.bim, b.fam)转为vcf文件
和正常plink文件类似,除了--file 变为--bfile即可。
plink --bfile b --recode vcf --out e
1.5 vcf 文件转化为plink文件(map和ped)
vcf 文件转化为plink文件map和ped
plink --vcf 1.vcf --recode --out b1.6 vcf 文件转化为plink文件(b.bed, b.bim, b.fam)
plink --vcf 1.vcf --out b- --vcf 需要文件名完整,不能只写前缀,所以这里要写
--vcf e.vcf - --recode 保存plink文件
功能之四:常用遗传参数
4.1 基因频率统计 : --freq 对所有位点进行基因频率统计
plink --file filename --freq --out maf

4.2 杂合度统计 : --het
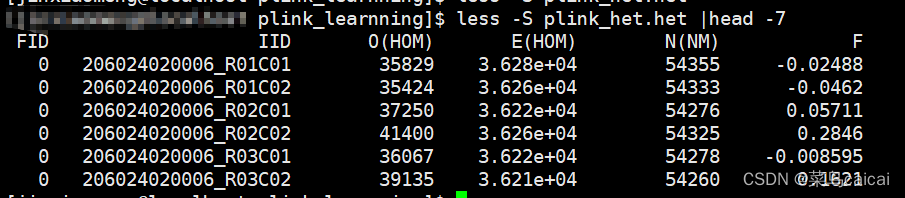
plink --file plink --het --out plink_het --chr-set 30
4.3 LD 计算两个SNP位点的连锁不平衡值: --ld
# output file : plink.ld
# 1. 分析一对SNP --ld
plink --bfile filename --ld snp1ID snp2ID
# 2. 多个SNP的LD分析 --r
plink --file mydata --r
# 3. 过滤输出
plink --bfile chr3 --ld-window 99999 --ld-window-kb 5000 --r2 --ld-window-r2 0.85 --out chr3
--bfile: 读取的基因型Bim文件
--ld-window 10 : 表达结果文件中输出变异最大数量
# 仅分析相距不超过10个SNP的SNP(默认为10个)
# 0 的话就是,所有标记两两间都计算到结果文件
--ld-window-kb 5000:计算一个位点周边5Mb范围点的LD # 对距离在5Mb之内的SNP位点进行分析(默认为1Mb)
--ld-window-r2 0.85: 保留r2 > 0.85的标记 查看日志剩余标记数
# .in剩余的标记 .out去除的标记 再用--extract提取.in出来 --exclue剔除.out中的位点
# 仅输出R2高于该参数值的LD分析结果(仅适用于--r2)
三、获取特定SNP与所有其他SNP的LD值
获取rs12345的1Mb内每个SNP的所有值的列表
plink --file mydata
--r2
--ld-snp rs12345
--ld-window-kb 1000
--ld-window 99999
--ld-window-r2 0
对于多个SNP,要从具有其他SNP的一组SNP中获取所有的LD值,命令为
# mysnps.txt是SNP列表
--ld-snp-list mysnps.txt
四、单倍体块估计
plink --bfile mydata --blocks
输出文件:plink.blocks,plink.blocks.det
# 此格式可与--hap命令一同使用,测试每个块中的每个单元型的关联性,或估算单元型的频率
# plink --bfile mydata --hap plink.blocks --hap-freq筛选SNP
SNP 数量太多,计算会非常慢。可以使用 plink 的 --indep-pairwise 命令,通过 LD 筛选位点:
plink --bfile data --indep-pairwise 50 10 0.1
plink --bfile data --extract plink.prune.in --make-bed --out data.pruned
这里的过滤参数的意思是:
50, 滑动窗口是50
10, 每次滑动的大小是10
0.1 表示R方小于0.1
#删除在50个SNP滑动窗口内(每次增加10个SNP)与任何其他SNP的R2值大于0.1的每个SNP。#结果文件 一对一对的
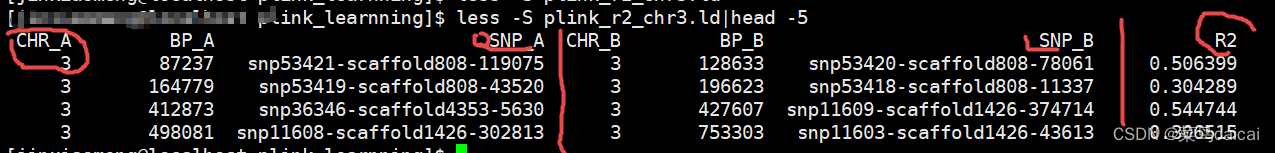
plink --file plink --r2 --ld-window-kb 5000 --chr 3 --out plink_r2_chr3 --chr-set 30
功能之五:计算亲缘关系IBS(2个个体遗传相似度)
IBD:需要有系谱数据但是也可以通过IBS推算;
IBS:用的多 --distance(plink)/
1.1 IBS:用的多 --distance(plink)/
plink --file filename --distance square ibs --out ibs
# --distance square ibs
# square 生成矩阵格式,下三角
# ibs 加此参数会输出IBS矩阵,1-ibs将会输出1-IBS的值,即遗传距离;不加默认输出等位基因数量表示的距离
#outfile .mibs/.mdist .mibs.id/.mdist.id #样本ID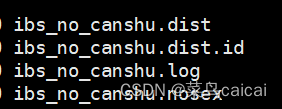
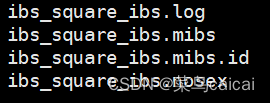
1.2 # 不加参数,不加默认输出等位基因数量表示的距离,下三角,无ID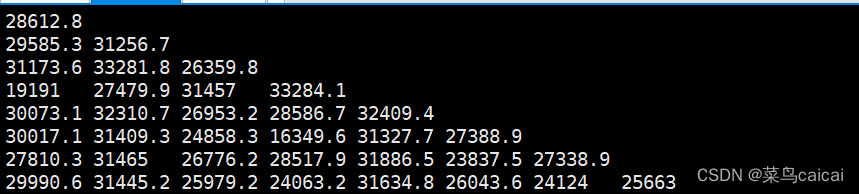
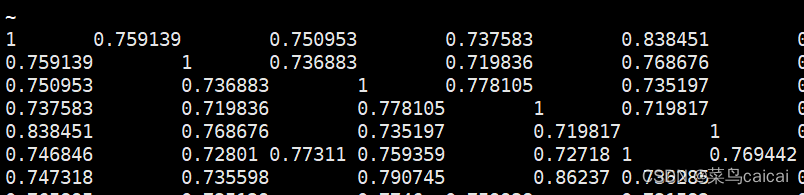
1.3 # 加参数 --distance square ibs,全部,对角线值为1 无ID,以矩阵形式表示
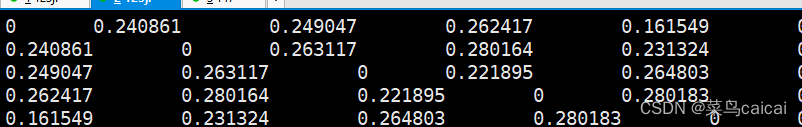
1.4 # 加参数--distance square 1-ibs 对角线为0,遗传距离的结果,样本1与样本1遗传距离为0
plink --file plink --distance square 1-ibs --out ibs/ibs_square_1_ibs --chr-set 30
1.5 # 加参数--genome,ibs, 有ID,DST ,以列表形式表示
plink --file plink --distance square ibs --genome --out ibs/ibs_square_ibs_genome --chr-set 30flat-missing 缺失矫正 Distance matrices - PLINK 1.9
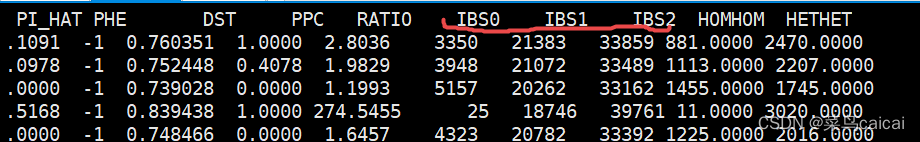
1.6 # 加参数--genom full 显示 多3列 IBS0 IBS1 IBS2
IBS0样本1与样本2全部等位基因不相同的位点的数量
DST=(IBS1+IBS2)/( IBS0+ IBS1+IBS2)
plink --file plink --distance square ibs --genome full --out ibs/ibs_square_ibs_genome_full --chr-set 30

2.1 PLINK计算IBD是根据位置和染色体信息
利用系谱信息估计亲缘系数为0.125时,反映的是至少3代以上个体间的亲缘关系。与此同时,有研究认为亲缘关系在3代以外的个体可以近似为无关个体。
--genome参数计算后得到PI_HAT(Proportion IBD)全是1。
# 利用IBD可以描述两个样本间的亲缘关系,采用plink计算(IBD)PI_HAT:
#该步骤进行样本间的亲缘关系估计,并将清除未标记明显亲子关系的个体亲缘关系过近的个体(GWAS)。
plink --file 3 --genome --out 4 --allow-no-sex --noweb
# 理想状态下父子关系的两个样本,Z0, Z1, Z2对应的值分别为0,1, 0,所有位点的一个allel都继承自父本;同卵双胞胎的两个样本,则为0,0,1,所有的allel都来自共同的祖先,对于异卵双胞胎,则为0.25,0.5,0.25
# PI_HAT这个统计量的取值范围为0-1,数值越大,两个样本的亲缘关系越近,当为1时,表示的就是同卵双胞胎,或者重复样本,可以根据这个值筛选亲缘关系近的样本进行过滤。
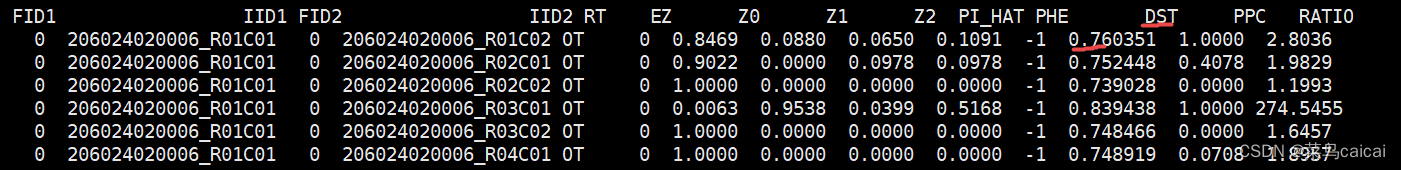
# 输出:4.genome
FID1 First sample’s family ID
IID1 First sample’s within-family ID
FID2 Second sample’s family ID
IID2 Second sample’s within-family ID
RT Relationship type inferred from .fam/.ped file,从fam/ped文件推断的样本关系 UN
EZ IBD sharing expected value, based on just .fam/.ped relationship NA
Z0 P(IBD=0)
Z1 P(IBD=1)
Z2 P(IBD=2)
PI_HAT P(IBD=2)+0.5*P(IBD=1) ( proportion IBD ) // Proportion IBD, i.e. P(IBD=2) + 0.5*P(IBD=1),IBD比例
PHE Pairwise phenotypic code (1,0,-1 = AA, AU and UU pairs) // Pairwise phenotypic code (1, 0, -1 = case-case, case-ctrl, and ctrl-ctrl pairs, respectively)
DST IBS distance (IBS2 + 0.5*IBS1) / ( N SNP pairs ) // IBS distance, i.e. (IBS2 + 0.5*IBS1) / (IBS0 + IBS1 + IBS2)
PPC IBS binomial test #二项式
RATIO Of HETHET : IBS 0 SNPs (expected value is 
PI_HAT值越大,亲缘关系越近,PI_HAT=1/2*Z1 + Z2,为1则可能为同卵双胞胎或者重复测序样本。Z0表示样本IID1与IID2的0个染色体组含有相同变异的频率,同理,z1/z2表示两个样本1个/2个染色体组含有相同变异频率。
这个IBD结果表明,227号与228号关系远,为两头母牛的测序号。
228与229关系更近,两样本Z1为0.6454,初步断定228为小牛229的母亲。当然还需PCA、系统树的辅助验证。
**实践证明:合并vcf文件计算得到的PI_HAT大的离谱,合并bam文件更合理。
使用SNP数据计算IBD、PCA、系统树鉴别亲缘关系_谁曾经不是菜鸟啦的博客-CSDN博客
无亲缘关系为何IBD结果为同卵双胞胎/重复样本_橙子牛奶糖的博客-CSDN博客
功能之六:计算亲缘关系构建G矩阵也可以用GCTA/plink
6.1. --make-grm(GCTA 算法两种,自选)----输出为二进制文件 :二进制
gcta --bfile filename --autosome --make-grm --make-grm-alg 0 --out kinship0
1 基于Van Raden
0 基于Yang
--autosome表示只使用常染色体上的SNP位点
test.grm.bin 这个是二进制存储的GRM
test.grm.N.bin 这个是二进制文件,存储的是参与计算的SNP个数
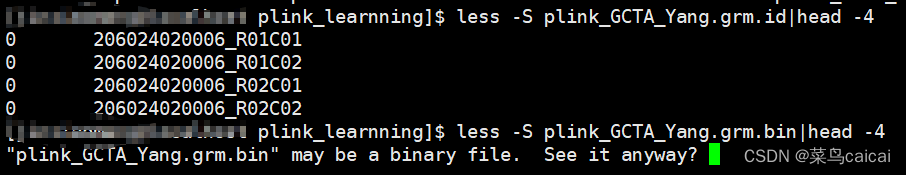
test.grm.id FID和IID的信息。
https://blog.csdn.net/yijiaobani/article/details/122437266
结果会生成矩阵的下三角,保存为二进制文件。
6.2 若还有后续分析,想查看样本间对应的亲缘关系,可以用以下方式,建议用 “--make-grm-gz”参数:文本形式
gcta64 --bfile test --make-grm-gz --make-grm-alg 1 --out kinship
eg:
/mnt/data//software/gcta_1.94.0beta/gcta64 --bfile plink --make-grm-gz --make-grm-alg 1 --out plink_GCTA_Vanrander
# --file 这个参数无效

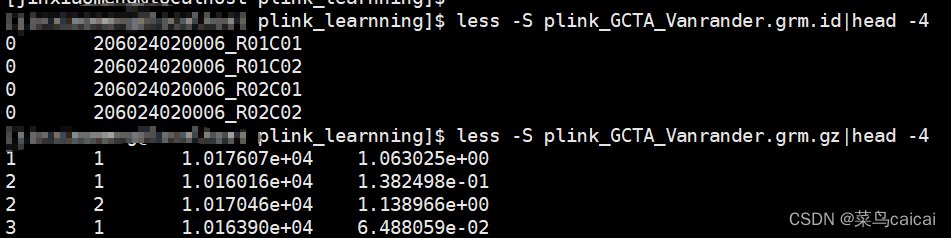
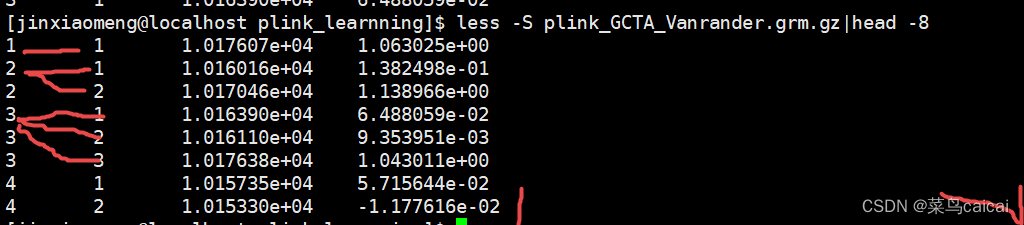
test.grm.gz (no header line; columns are indices of pairs of individuals (row numbers of the test.grm.id), number of non-missing SNPs and the estimate of genetic relatedness)
第一列和第二列为编号信息(根据ID的顺序编号,相当于是矩阵的下三角行列信息),
第三列表示两个样本中都分型成功的SNP位点个数,
第四列表示两个样本间的亲缘关系值
test.grm.id (no header line; columns are family ID and individual ID) 为FID和IID数据,第一列为家系信息,第二列是个体ID。
2. --make-rel square (plink 算法基于Yang)
plink --file filename --make-rel square
plink --file plink --make-rel --out G/plink_ral --chr-set 30
#square 生成矩阵格式,默认生成下三角矩阵
# .rel .rel.id
#对角线的值接近1 #无ID,需要和id整合 可热图可视化 下三角矩阵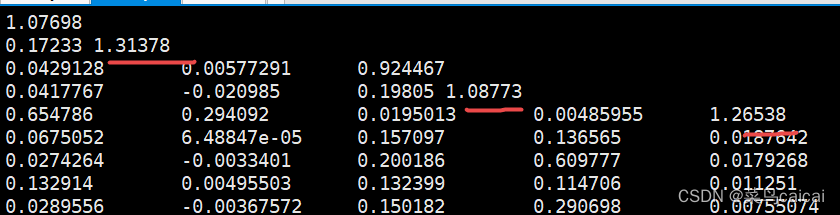
功能之七:计算近交系数 --homozyg 统计ROH
在基因组某一段区域内,当一定数量一定密度的 SNPs 表现为纯合时,可以判断该区域存在 ROHs 现象
PLINK 中使用滑动窗口的方法来进行 ROHs 的检测:特定 SNP 数目的窗口以一定的步长在 SNP 芯片上滑动
# --homozyg 计算ROH(连续性纯合片段(runs of homozygosity, ROH))
plink --bfile bsz --homozyg --allow-extra-chr
plink --file ROH --homozyg-snp 30 --homozyg-kb 1000 --homozyg-density 1000 --homozyg-gap 1000 --homozyg-window-snp 50 --homozyg-window-het 1 --homozyg-window-missing 1
示例代码如下所示:
plink \
--bfile ${input} \
--homozyg \
--homozyg-density 1000 \ 一段ROH中每1000kb必须有1个SNP
--homozyg-gap 1000 \ 如果连续两个SNP的间隔大于1000kb,那么就不能归为同一个ROH;认定相邻ROH
--homozyg-kb 500 \ 只检测长度大于500kb的ROH
--homozyg-snp 50 \ 只检测长度超过50个SNP的ROH
# --homozyg-snp 30 --homozyg-kb 仅记录包含至少30个SNPs且总长度≥ 1000千个碱基的纯合性
--homozyg-window-het 1 \ ROH滑窗中可以允许有一个SNP位点为杂合
--homozyg-window-missing 1 默认情况下,扫描窗口命中最多可以包含1个杂合调用和1个缺失调用;
--homozyg-window-snp 50 \ 滑窗大小为50个SNP
--homozyg-window-threshold 0.05 \ 包含某个SNP的完全纯合滑窗的比例至少为5%
--out ${output}计算完成后会得到 .hom 与.hom.indiv 等文件

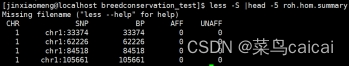
.hom 文件包含了每段ROH的具体信息
FID IID PHE CHR SNP1 SNP2 POS1 POS2 KB NSNP DENSITY PHOM PHET
1 1 -9 chr1 rsxxx rsxxx 12345 67891 5.5 123 9.17 0.99 0.07
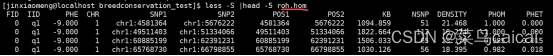
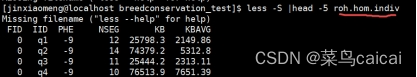
hom.indiv文件则是包含了对每个个体ROH汇总信息,包括总长度与总个数
FID IID PHE NSEG KB KBAVG
1 1 -9 50 75000 1500
统计ROHs和计算近交系数
近交系数 (FROH) =
![]()
Plink常用命令总结_Taylent的博客-CSDN博客_plink
功能之八:PCA --pca
6.1 利用plink计算pca-----直接出结果
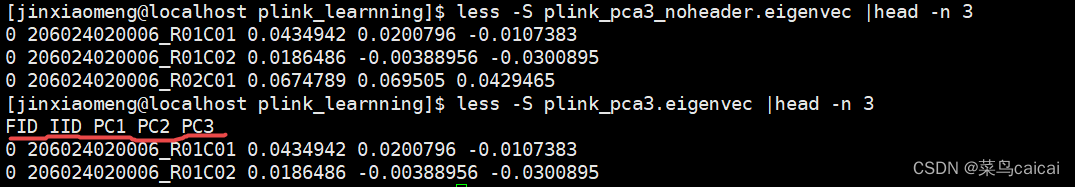
plink --file plink --pca 3 header --out plink_pca
#如
plink --file plink --pca 3 header --out plink_pca3 --chr-set 30
#默认是提取前20个pca
# header 结果文件加表头,默认没有表头
out.file 特征值.eigenval .eigenvec
#解释的方差 (PCA1/(PCA1 + PCA2 +PCA3)) 
结果文件具体内容

6.2 利用GCTA计算pca-----分2步;先计算亲缘关系矩阵---再计算PCA
1.转bed格式
# 因为我没有fam文件,用plink转换二进制文件
# 省略 plink --file plink --make-bed --out plink --chr-set 30
2.计算亲缘关系
gcta_1.94.0beta/gcta64 --bfile plink --make-grm --make-grm-alg 0 --out plink_GCTA_Yang
# GCTA主要是分析人的数据,如果分析其他动植物需要对染色体体进行重新编号,不然会报错:
Error: Line 56287 of [plink.bim] contains illegal chr number, please check
主要做人的 要求为数字染色体:
#用plink提取染色体 plink --file plink --make-bed --out plink --chr-set 30 --chr 1-13
3.pca
gcta_1.94.0beta/gcta64 --grm plink_GCTA_Yang --pca 3 --out plink_GCTA_Yang_pca3 
功能之九:基因型形势转换 0/1/2 1/2/3/4 A/T/C/G
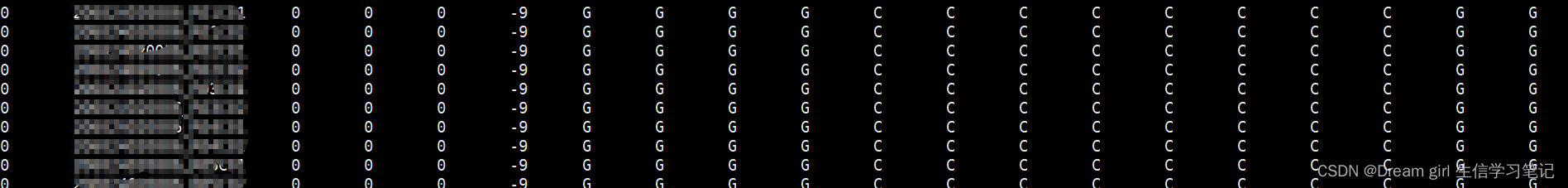
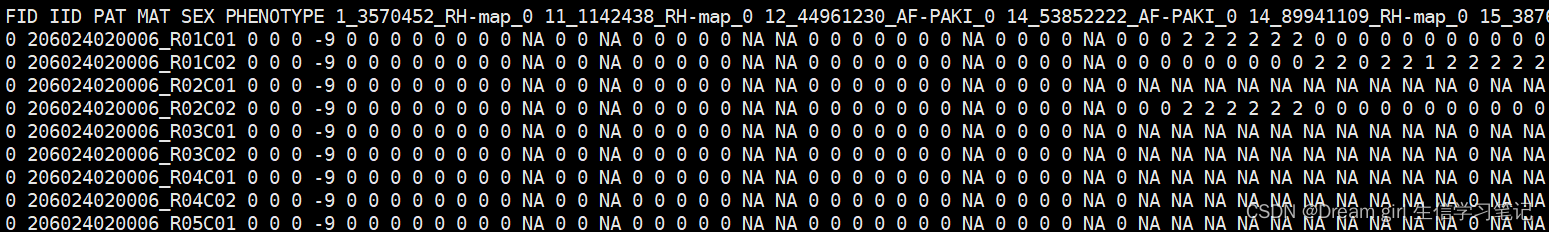
plink --file plink --recode A --out plink_allele012 --chr-set 50
--recode A: 数字格式,0和2纯合 ,1 杂合
plink --file plink --allele1234 --recode tabx --out
--allele1234 : A/T/C/G表示为1/2/3/4
--alleleACGT: 表示为A/C/G/T
plink --file plink --recode A --out plink_allele012 --chr-set 50
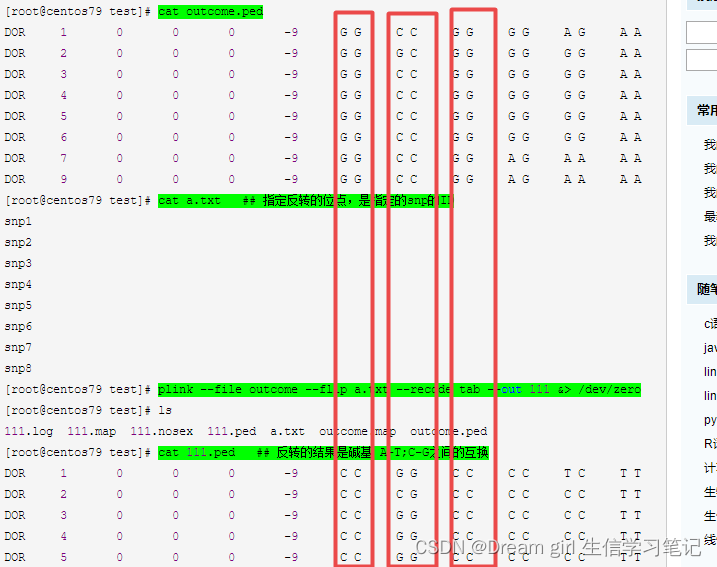
参考:plink --flip 命令实现正负链的反转 - 美洲豹2018 - 博客园
本文章仅作为个人笔记与大家分享,希望大家少走弯路,过程中参考了一系列大神的文章,如有侵犯劳烦联系删除。
刚刚探索微信订阅号,欢迎大家关注,会不定时分享农学相关生信分析相关流程。


















 2590
2590

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









