






一、写在前面
现在的单细胞市场,早就不是10下、BD瓜分天下的年代了,越来越多性能不俗的国产平台走上饭桌。多种多样的单细胞测序平台也给大家带来了过剩的备选方案,此时如何选择适合自己组织类型/研究目的需求的单细胞平台就成了一个难题。毕竟单细胞也是种单样本接近万元级的技术,立项前对平台的评估与选择显得尤为重要。这里我们收集了来源于三个单细胞平台、年份相近的肺部组织数据,从nCount、nFeature、分群注释效果等方面对这些平台的数据进行了评估。希望对大家后续选择单细胞平台提供一些启示,大家立项前也可以挑选自己熟悉平台与组织进行测试。如果你看不懂以下分析流程,那么你需要学习:不知不觉,已分享了近两百篇单细胞知识帖
本教程基于Linux中的Rstudio环境演示,计算资源不足的同学可参考:
生信分析为什么要使用服务器?
足够支持你完成硕博生涯的生信环境
配置一个心仪的工作站(硬件+环境配置)
独享服务器,生信分析不求人
为实验室准备一份生物信息学不动产
二、数据准备
1、环境设置
setwd("/home/cwj/project/08_depth_compar/")
suppressMessages({
library(Seurat)
library(Matrix)
library(ggplot2)
library(patchwork)
library(magrittr)
})2、下载、解压及格式处理
(1) 10x
来源:https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE266323物种:Mus musculus
组织:肺
公布时间:2024
(2) BD
来源:Apelin modulates inflammation and leukocyte recruitment in experimental autoimmune encephalomyelitis物种:Mus musculus
组织:肺
公布时间:2024
cd /home/cwj/project/08_depth_compar/data/GSE230551_RAW
for i in`ls /home/cwj/project/08_depth_compar/data/GSE230551_RAW`;do
gzip -d $i
done(3) SeekGene
来源:https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE283551物种:Mus musculus
组织:肺
公布时间:2025
rm -rf /home/cwj/project/08_depth_compar/data/GSE283551_RAW
mkdir -p "GSE283551_RAW"&& tar -xf "GSE283551_RAW.tar"-C "GSE283551_RAW"
cd /home/cwj/project/08_depth_compar/data/GSE283551_RAW
for i in C73 C80 E19 E27;do
mkdir $i
mv /home/cwj/project/08_depth_compar/data/GSE283551_RAW/*${i}*barcodes.tsv.gz /home/cwj/project/08_depth_compar/data/GSE283551_RAW/$i/barcodes.tsv.gz
mv /home/cwj/project/08_depth_compar/data/GSE283551_RAW/*${i}*features.tsv.gz /home/cwj/project/08_depth_compar/data/GSE283551_RAW/$i/features.tsv.gz
mv /home/cwj/project/08_depth_compar/data/GSE283551_RAW/*${i}*matrix.mtx.gz /home/cwj/project/08_depth_compar/data/GSE283551_RAW/$i/matrix.mtx.gz
done3、数据读入
(1) 10x
dir <-list.dirs("/home/cwj/project/08_depth_compar/data/GSE266323_RAW")[-1]
names(dir) <-c("d101","d102","dTom1","dTom2")
dir
obj_list <-list()
for(i in1:length(dir)){
counts <-Read10X(data.dir = dir[i])
obj_list[[i]] <-CreateSeuratObject(counts,project =names(dir)[i],min.cells=0,min.features=0)
}
obj_list
obj_10x <-merge(x=obj_list[[1]],y=obj_list[-1])
obj_10x <-JoinLayers(obj_10x)
obj_10x
table(obj_10x@meta.data$orig.ident)(2) BD
names(dir) <-c("Cont","EAE-Cont","EAE-A13")
dir
obj_list <-list()
for(i in1:length(dir)){
counts <-read.table(paste0("/home/cwj/project/08_depth_compar/data/GSE230551_RAW/",dir[i]),header = T)
print(head(counts))
colnames(counts)[2] <-"Gene"#有的基因列名是"Bioproduct"
sparse_matrix <-sparseMatrix(
i =as.numeric(factor(counts$Gene)),
j =as.numeric(factor(counts$Cell_Index)),
x = counts$RSEC_Adjusted_Molecules
)
# 将行和列名称设置为基因名称和细胞索引
rownames(sparse_matrix) <-levels(factor(counts$Gene))
colnames(sparse_matrix) <-levels(factor(counts$Cell_Index))
obj_list[[i]] <-CreateSeuratObject(sparse_matrix,project =names(dir)[i],min.cells=0,min.features=0)
}
#BD平台得到的矩阵文件,features.tsv.gz中基因数目并不是固定的,即每个样本基因数目不同
obj_list
obj_BD <-merge(x=obj_list[[1]],y=obj_list[-1])
obj_BD <-JoinLayers(obj_BD)
obj_BD
table(obj_BD@meta.data$orig.ident)(3) SeekGene
三、数据质量展示
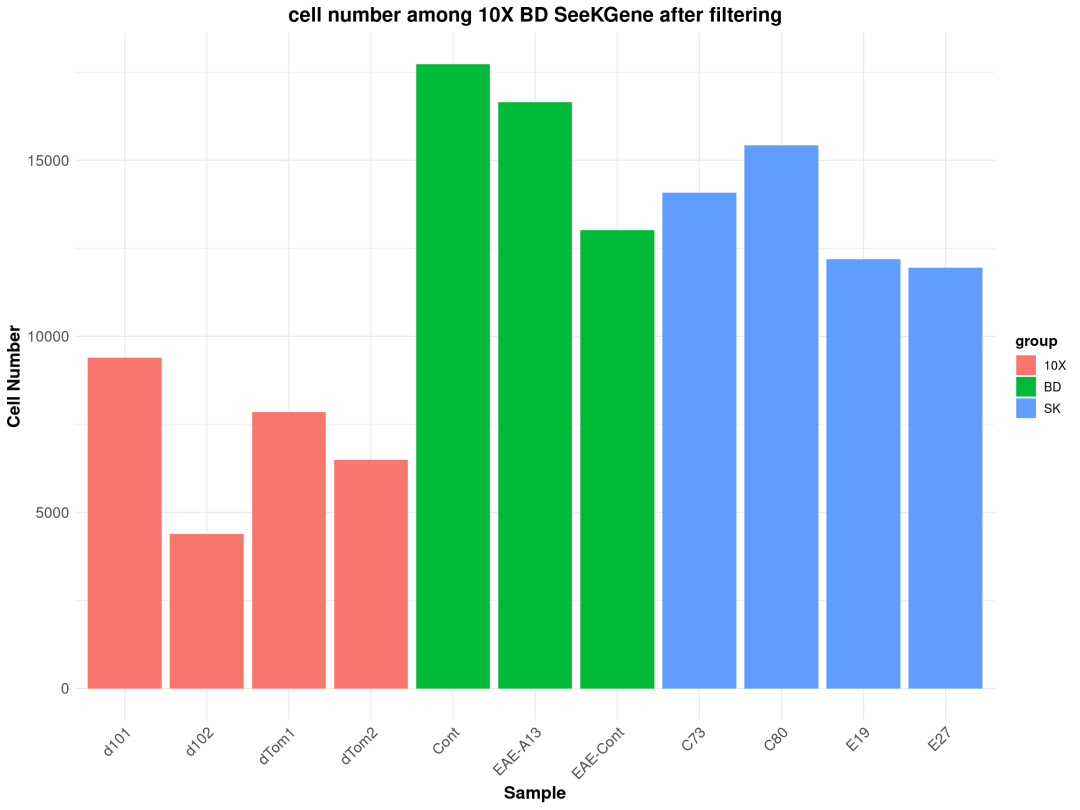
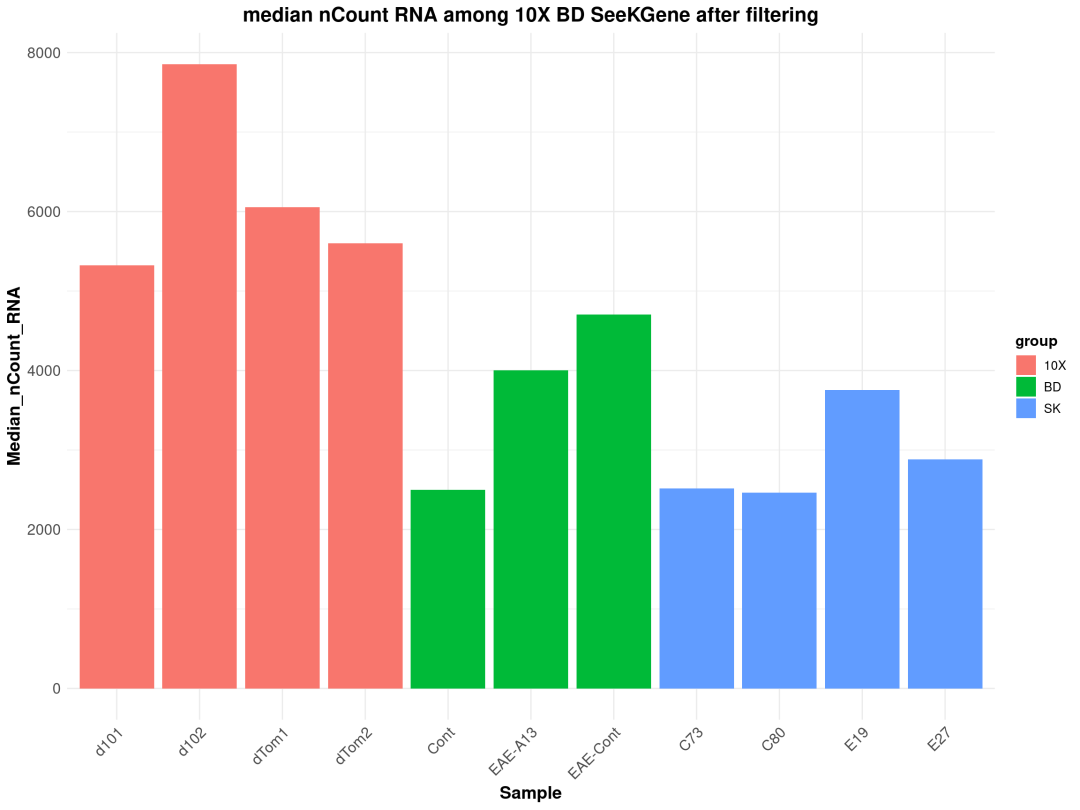
1、肺组织质量展示
ggplot(lung_summary_after_filtering, aes(x = Cell_Count.Var1 , y = Median_nFeature_RNA, fill= group)) +geom_bar(stat ="identity")+
labs(x ="Sample", y ="Median_nFeature_RNA", title ="median nFeature RNA among 10X BD SeeKGene after filtering") +
theme_minimal() +
theme(axis.text.x =element_text(angle =45, hjust =1, size =12),
axis.text.y =element_text(size =12),
axis.title =element_text(size =14, face ="bold"),
plot.title =element_text(size =16, face ="bold", hjust =0.5),
legend.title =element_text(size =12, face ="bold"),
legend.text =element_text(size =10)) 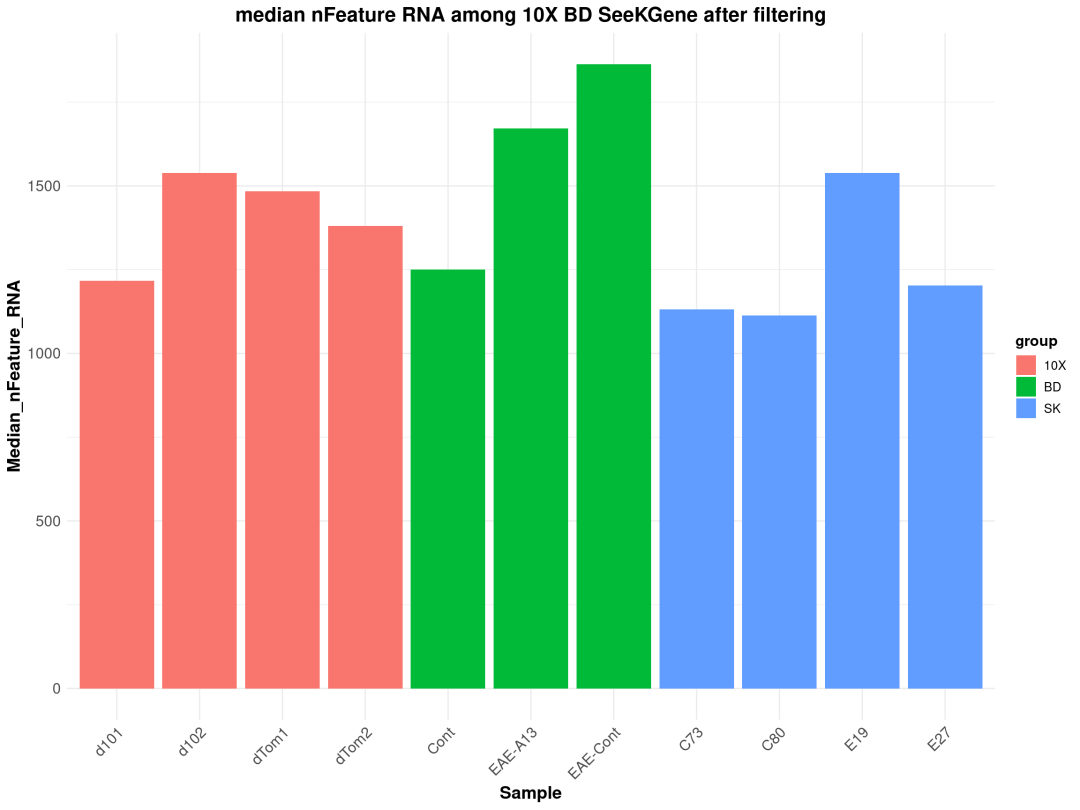
2、10x、BD、SeeKGene降维分群
# 运行时间较长,可以将结果运行一次后保存rds,后续直接读取
# obj_10x_filter <- readRDS("/home/cwj/project/08_depth_compar/result/obj_10x_filter.rds")
# obj_10x_res1.0<- obj_10x_filter %>%
# NormalizeData() %>%
# FindVariableFeatures() %>%
# ScaleData() %>% RunPCA() %>%
# FindNeighbors() %>%
# FindClusters(resolution = 1.0) %>%
# RunTSNE(dims = 1:10) %>%
# RunUMAP(dims = 1:10)
#saveRDS(obj_10x_res1.0, "/home/cwj/project/08_depth_compar/result/obj_10x_filter_res1.0.rds")
obj_10x_res1.0<-readRDS("/home/cwj/project/08_depth_compar/result/obj_10x_filter_res1.0.rds")
DimPlot(obj_10x_res1.0, reduction ='umap', label=T) +NoLegend() +NoAxes() 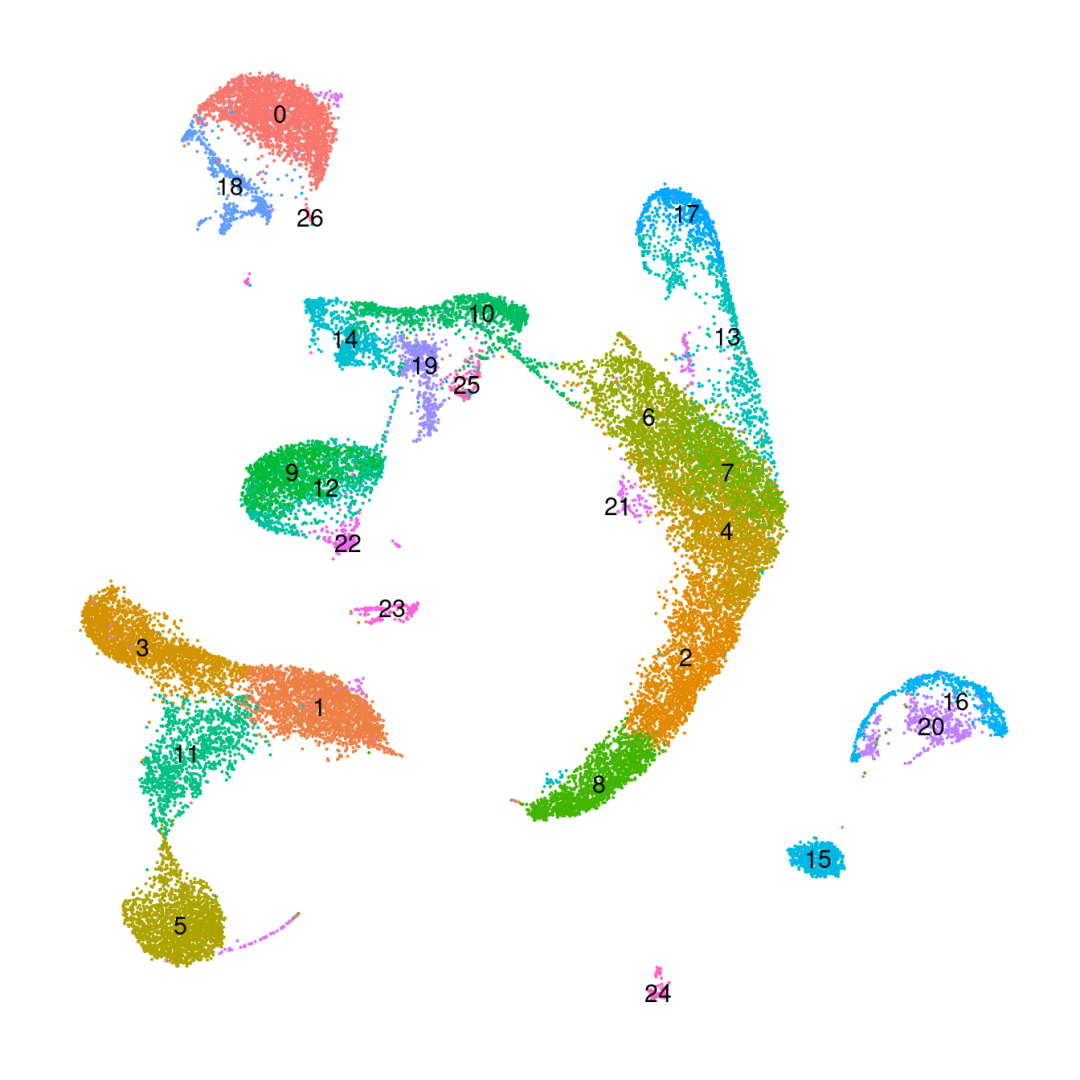
# obj_BD_filter <- readRDS("/home/cwj/project/08_depth_compar/result/obj_BD_filter.rds")
# obj_BD_res1.0 <- obj_BD_filter %>%
# NormalizeData() %>%
# FindVariableFeatures() %>%
# ScaleData() %>% RunPCA() %>%
# FindNeighbors() %>%
# FindClusters(resolution = 1.0) %>%
# RunTSNE(dims = 1:10) %>%
# RunUMAP(dims = 1:10)
#saveRDS(obj_BD_res1.0, "/home/cwj/project/08_depth_compar/result/obj_BD_filter_res1.0.rds")
obj_BD_res1.0<-readRDS("/home/cwj/project/08_depth_compar/result/obj_BD_filter_res1.0.rds")
DimPlot(obj_BD_res1.0, reduction ='umap', label=T) +NoLegend() +NoAxes() 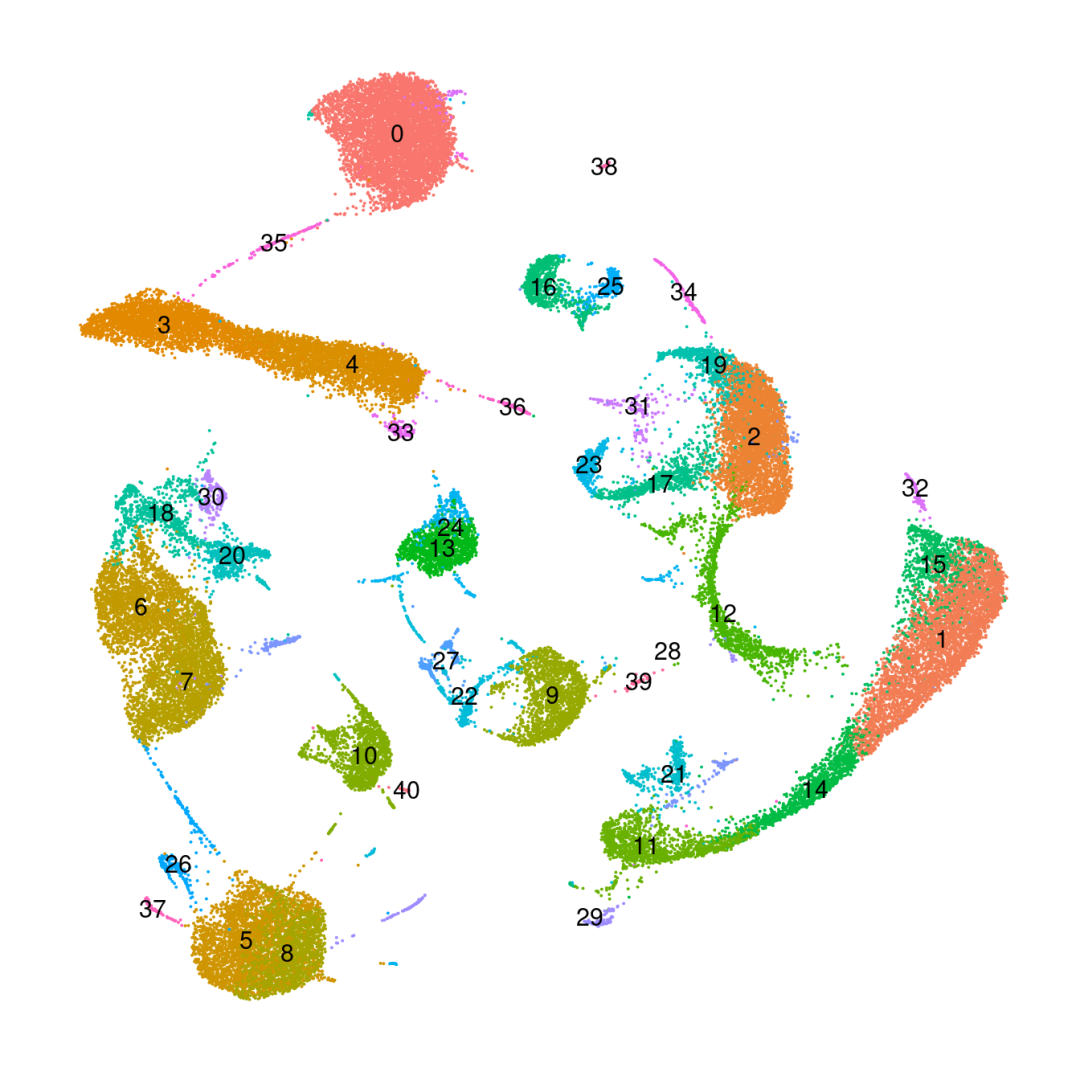
# obj_SK_filter <- readRDS("/home/cwj/project/08_depth_compar/result/obj_SK_filter.rds")
# obj_SK_res1.0 <- obj_SK_filter %>%
# NormalizeData() %>%
# FindVariableFeatures() %>%
# ScaleData() %>% RunPCA() %>%
# FindNeighbors() %>%
# FindClusters(resolution = 1.0) %>%
# RunTSNE(dims = 1:10) %>%
# RunUMAP(dims = 1:10)
#saveRDS(obj_SK_res1.0, "/home/cwj/project/08_depth_compar/result/obj_SK_filter_res1.0.rds")
obj_SK_res1.0<-readRDS("/home/cwj/project/08_depth_compar/result/obj_SK_filter_res1.0.rds")
DimPlot(obj_SK_res1.0, reduction ='umap', label=T) +NoLegend() +NoAxes() 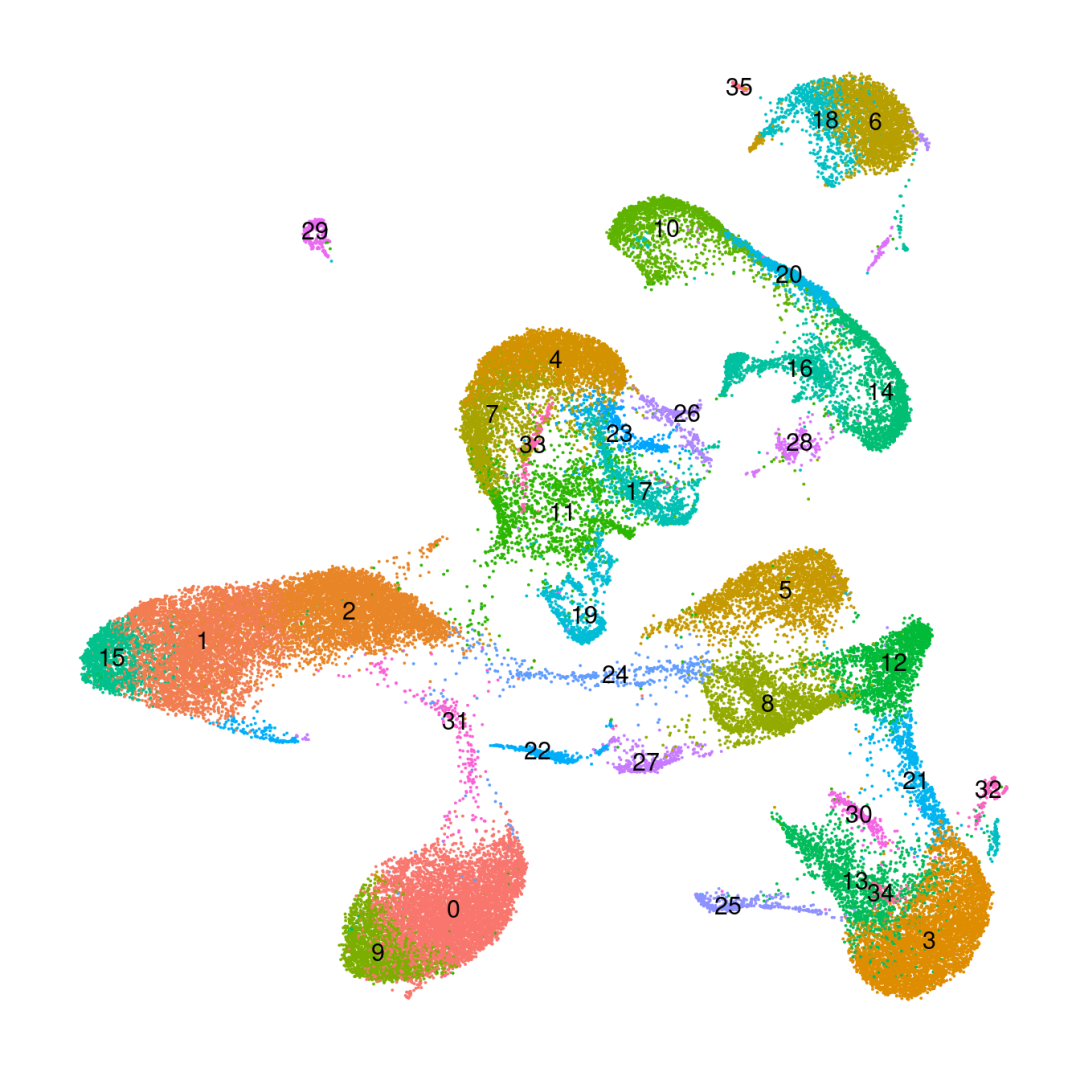
FeaturePlot(obj_10x_res1.0, features=c("nFeature_RNA","nCount_RNA","percent.mt"),ncol =3)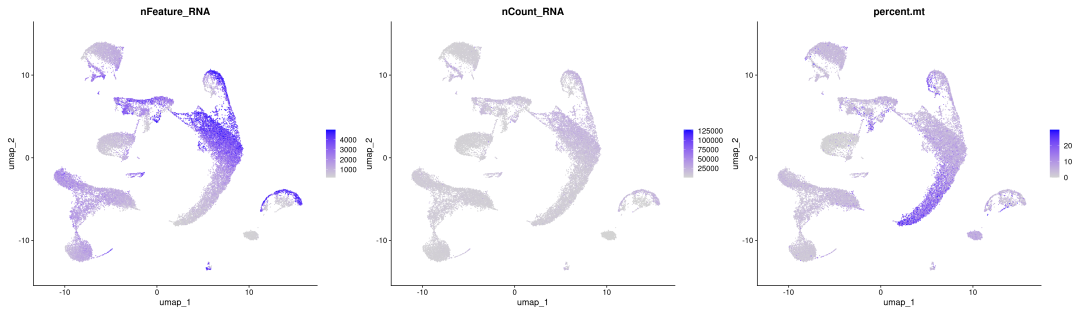
FeaturePlot(obj_BD_res1.0, features=c("nFeature_RNA","nCount_RNA","percent.mt"),ncol =3)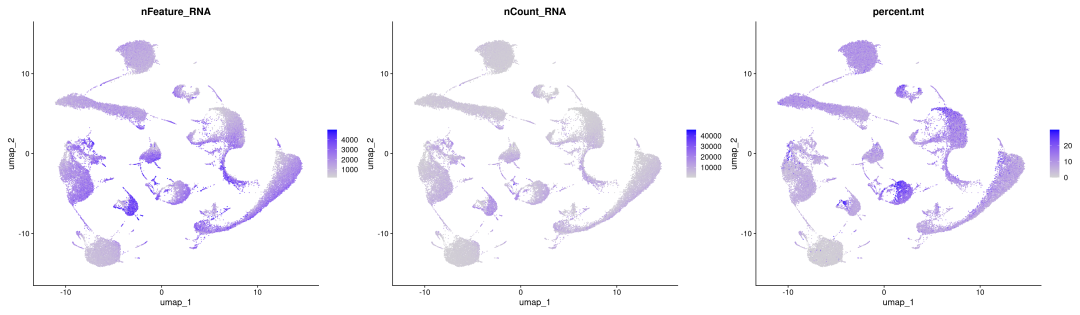
FeaturePlot(obj_SK_res1.0, features=c("nFeature_RNA","nCount_RNA","percent.mt"),ncol =3)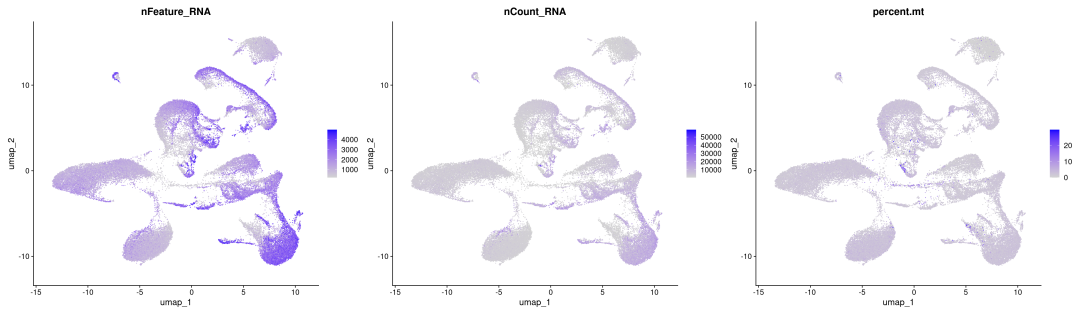
四、最后聊聊
粗暴地看上文分析结果,在细胞数量上BD与SeekGene完胜10X,nFeature数的性能三家相差无几,nCount数上10X则是遥遥领先;在降维分群方面,在同样1.0的分辨率下10X的cluster类型最少,BD与SeekGene获得的cluster更多,但BD似乎出现了降维拖尾的现象,过滤参数还需要进一步调整。当然,这只是一个非常粗糙的评估,我们的小测评并不能考虑到数据来源、建库、测序完成度的一致性,也未能对大队列的数据进行横向评估,并且仅限于肺部一个组织,所以我们这里的结果仅供大家参考(保命申明)。


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









