






一、写在前面
此前我们也分享过类似功能的R包,与Rcircos将信息花在圆环上不同,RIdeogram可用于利用基因组相关数据在染色体上绘制相关信息,文章收录在Hao Z, Lv D, Ge Y, Shi J, Weijers D, Yu G, Chen J. 2020. RIdeogram: drawing SVG graphics to visualize and map genome-wide data on the idiograms. PeerJ Comput. Sci. 6:e251。总体上来说这个包的用法很简单,其只包含三个函数:ideogram、convertSVG、GFFex。可视化集锦可见:

二、图文教程
# 安装包:
install.packages("devtools")
devtools::install_github('TickingClock1992/RIdeogram')library(RIdeogram)
## Warning: 程辑包'RIdeogram'是用R版本4.3.3 来建造的一、测试数据
如果以下数据你都无法正常加载,可以先加载一下我保存的镜像:
load('biomamba.rdata')我们来看一下这个包内置的几个测试数据:karyotype数据如同他的名字一样,包含了染色体核型数据,一共包含五列,第一列是染色体ID,第二列和第三列是对应染色体的起始位点和终止位点,第三列和第四列是染色体对应着丝粒的起始位点和终止位点(最后两列着丝粒信息可以省略,必要的是前三列)。
data(human_karyotype, package="RIdeogram")
head(human_karyotype)## Chr Start End CE_start CE_end
## 1 1 0 248956422 122026459 124932724
## 2 2 0 242193529 92188145 94090557
## 3 3 0 198295559 90772458 93655574
## 4 4 0 190214555 49712061 51743951
## 5 5 0 181538259 46485900 50059807
## 6 6 0 170805979 58553888 59829934gene_density这里就是各位想要绘制的信息(mydata),一般包含四列,第一列为染色体ID,第二列和第三列是对应数据(例如你做的是WGBS的DMR差异分析,那这里的起始位点就是对应DMR窗口的起始位点)的起始位点和终止位点,第四列代表的就是这个位点对应的填充值(这里是对应区段的基因密度,如果拿WGBS举例,那就是甲基化的差异倍数)
data(gene_density, package="RIdeogram")
head(gene_density)## Chr Start End Value
## 1 1 1 1000000 65
## 2 1 1000001 2000000 76
## 3 1 2000001 3000000 35
## 4 1 3000001 4000000 30
## 5 1 4000001 5000000 10
## 6 1 5000001 6000000 10# 基因密度文件也可以由GFF文件自动生成:
download.file('ftp://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/release_32/gencode.v32.annotation.gff3.gz','gencode.v32.annotation.gff3.gz')
gene_density <- GFFex(input = "gencode.v32.annotation.gff3.gz",# gff注释文件
karyotype = "human_karyotype.txt", feature = "gene", # 以基因一列计算密度
window = 1000000)# 计算基因密度的窗口大小Random_RNAs_500文件主要记录标签信息(mydata_interval),这里的测试文件共包含六列;第一列为标签类型,第二列是标签对应的形状,第三列是染色体名称,第四列和第五列是对应数据在染色体上的起始位点和终止位点,第六列是希望标记的对应颜色。
data(Random_RNAs_500, package="RIdeogram")
head(Random_RNAs_500)## Type Shape Chr Start End color
## 1 tRNA circle 6 69204486 69204568 6a3d9a
## 2 rRNA box 3 68882967 68883091 33a02c
## 3 rRNA box 5 55777469 55777587 33a02c
## 4 rRNA box 21 25202207 25202315 33a02c
## 5 miRNA triangle 1 86357632 86357687 ff7f00
## 6 miRNA triangle 11 74399237 74399333 ff7f00当然,以上文件你也可以自己DIY制作,只要保证各文件的染色体名称、起始位点能够一一对应的上即可。
二、染色体图绘制
ideogram便是RIdeogram的核心可视化函数,遗憾的是这个函数不能将图片直接输出到Rstudio的画布中,而是输出到本地svg图片:
ideogram(karyotype = human_karyotype,# 染色体"底图"
output = "chromosome_1.svg")
# 将svg图片转换为png:
convertSVG("chromosome_1.svg", device = "png",file = 'chromosome_1.png')## Warning in checkValidSVG(doc, warn = warn): This picture may not have been
## generated by Cairo graphics; errors may result
RIdeogram默认生成svg图片
svg2tiff("chromosome.svg",# 输入图片路径
file = 'output.tiff', # 输出图片路径
dpi = 300# 输出图片质量
)
svg2pdf("chromosome.svg",# 输入图片路径
file = 'output.pdf', # 输出图片路径
dpi = 300# 输出图片质量
)
svg2jpg("chromosome.svg",# 输入图片路径
file = 'output.jpg', # 输出图片路径
dpi = 300# 输出图片质量
)
svg2png("chromosome.svg",# 输入图片路径
file = 'output.png', # 输出图片路径
dpi = 300# 输出图片质量
)就像我们上面说的,human_karyotype可以仅包含三列,着丝粒信息不是必须的:
ideogram(karyotype = human_karyotype[,1:3],# 染色体"底图"
output = "chromosome_2.svg")
# 将svg图片转换为png:
convertSVG("chromosome_2.svg", device = "png",,file = 'chromosome_2.png')## Warning in checkValidSVG(doc, warn = warn): This picture may not have been
## generated by Cairo graphics; errors may result
# 把基因密度以热图的形式绘制在染色体上:
ideogram(karyotype = human_karyotype, overlaid = gene_density,output = 'chromosome_genedensity_3.svg')
convertSVG("chromosome_genedensity_3.svg", device = "png",file = 'chromosome_genedensity_3.png')## Warning in checkValidSVG(doc, warn = warn): This picture may not have been
## generated by Cairo graphics; errors may result在上图中,可以进一步加入大家想要的标签,这里加入的是RNA的类型:
ideogram(karyotype = human_karyotype, label = Random_RNAs_500, label_type = "marker",output = 'chromosome_label_4.svg')
convertSVG("chromosome_label_4.svg", device = "png",file = 'chromosome_label_4.png')## Warning in checkValidSVG(doc, warn = warn): This picture may not have been
## generated by Cairo graphics; errors may result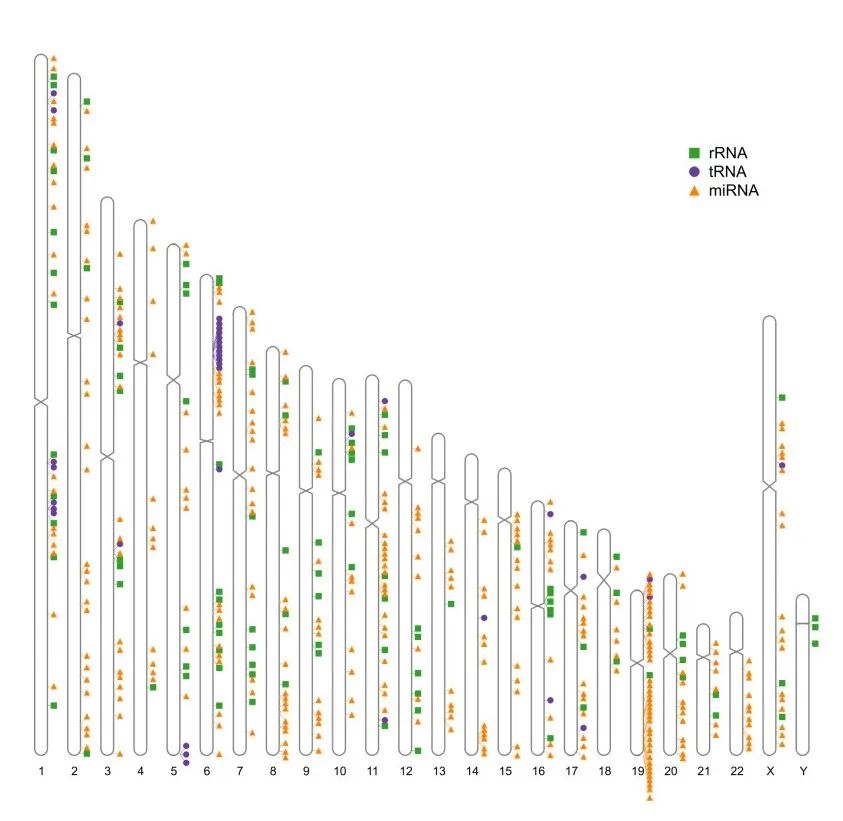
热图和自定义标签可以同时在ideogram中生效:
ideogram(karyotype = human_karyotype, overlaid = gene_density, label = Random_RNAs_500, label_type = "marker",output = 'chromosome_5.svg')
convertSVG("chromosome_5.svg", device = "png",file = 'chromosome_5.png')## Warning in checkValidSVG(doc, warn = warn): This picture may not have been
## generated by Cairo graphics; errors may result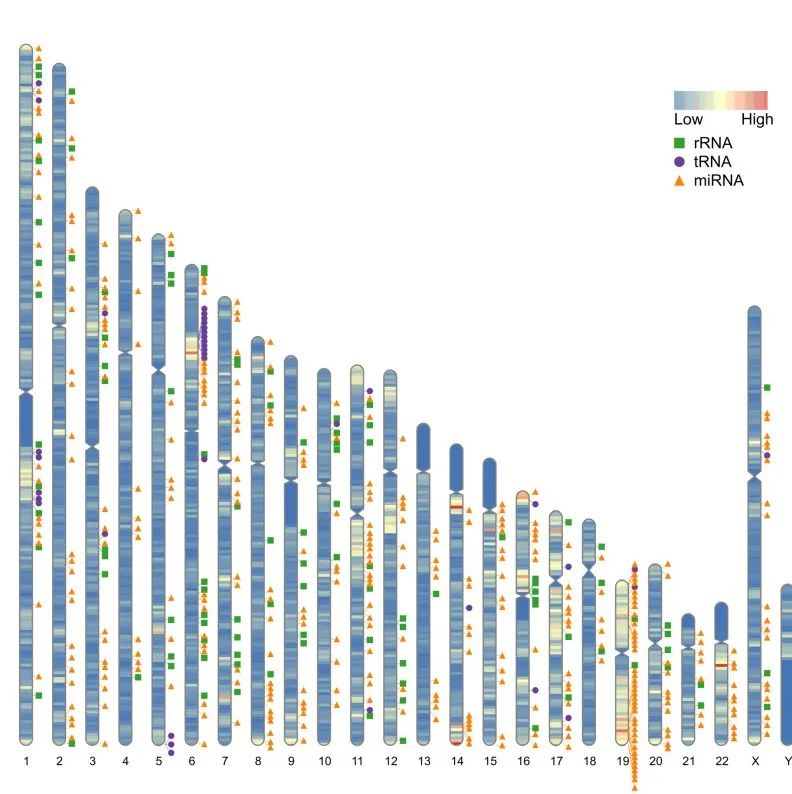
热图的颜色范围也可以自定义:
ideogram(karyotype = human_karyotype, overlaid = gene_density, label = Random_RNAs_500, label_type = "marker",
colorset1 = c("#fc8d59", "#ffffbf", "#91bfdb"# 自定义颜色
),output='chromosome_6.svg')
convertSVG("chromosome_6.svg", device = "png",file = 'chromosome_6.png')## Warning in checkValidSVG(doc, warn = warn): This picture may not have been
## generated by Cairo graphics; errors may result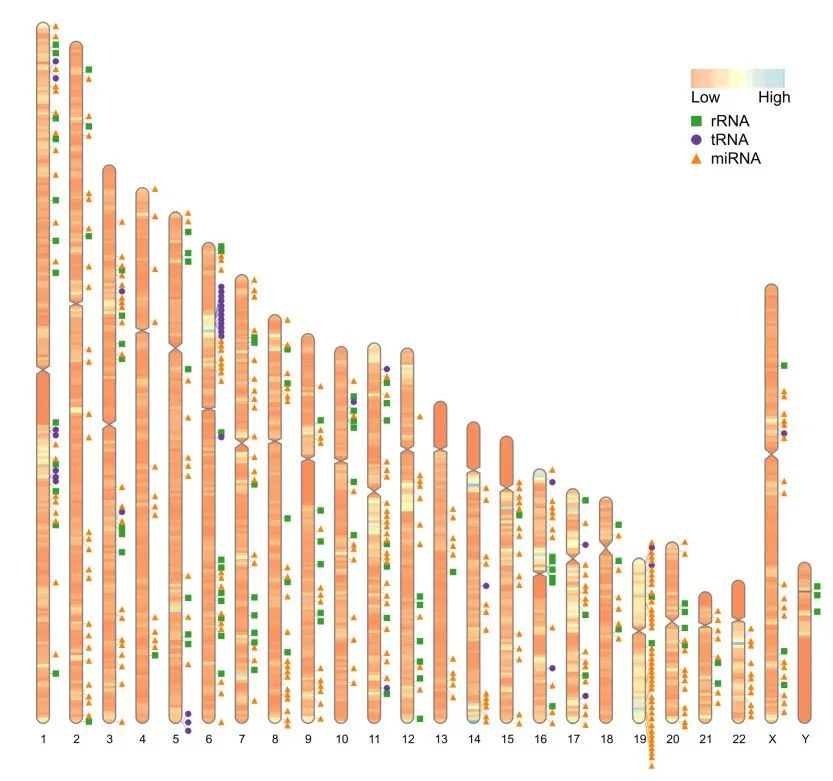
有时你可能只希望展示部分的染色体,或者说你的物种只有部分的染色体,那么你可以过滤数据并调整图片宽度:
library(dplyr)## Warning: 程辑包'dplyr'是用R版本4.3.3 来建造的##
## 载入程辑包:'dplyr'## The following objects are masked from 'package:stats':
##
## filter, lag## The following objects are masked from 'package:base':
##
## intersect, setdiff, setequal, union# 取出染色体1~10的数据:
filter_human_karyotype <- human_karyotype %>% filter(Chr %in% 1:10)
# 调整宽度前:
ideogram(karyotype = filter_human_karyotype, overlaid = gene_density, label = Random_RNAs_500,
label_type = "marker",output = 'chromosome_7.svg')
convertSVG("chromosome_7.svg", device = "png",file = 'chromosome_7.png')## Warning in checkValidSVG(doc, warn = warn): This picture may not have been
## generated by Cairo graphics; errors may result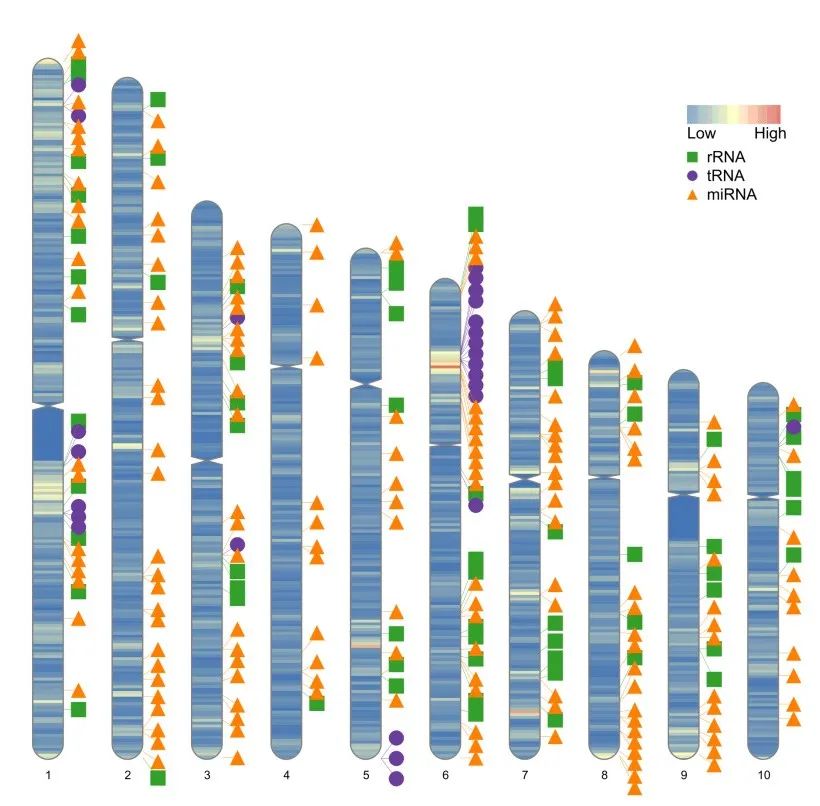
ideogram(karyotype = filter_human_karyotype,# 过滤数据,后面gene_density与Random_RNAs_500中的数据也会自动与human_karyotype对齐
overlaid = gene_density, label = Random_RNAs_500, label_type = "marker", width = 100,output = 'chromosome_8.svg')
convertSVG("chromosome.svg", device = "png",file = 'chromosome_8.png')## Warning in checkValidSVG(doc, warn = warn): This picture may not have been
## generated by Cairo graphics; errors may result
如果你希望调整标签的位置,可以通过Lx和Ly来完成,这两个值分别是与左上角位置的X方向、Y方向距离
ideogram(karyotype = human_karyotype, overlaid = gene_density, label = Random_RNAs_500, label_type = "marker", width = 100,
Lx = 80, # X方向的距离
Ly = 25, # Y方向的距离
output = 'chromosome_9.svg')
convertSVG("chromosome_9.svg", device = "png",file = 'chromosome_9.png')## Warning in checkValidSVG(doc, warn = warn): This picture may not have been
## generated by Cairo graphics; errors may result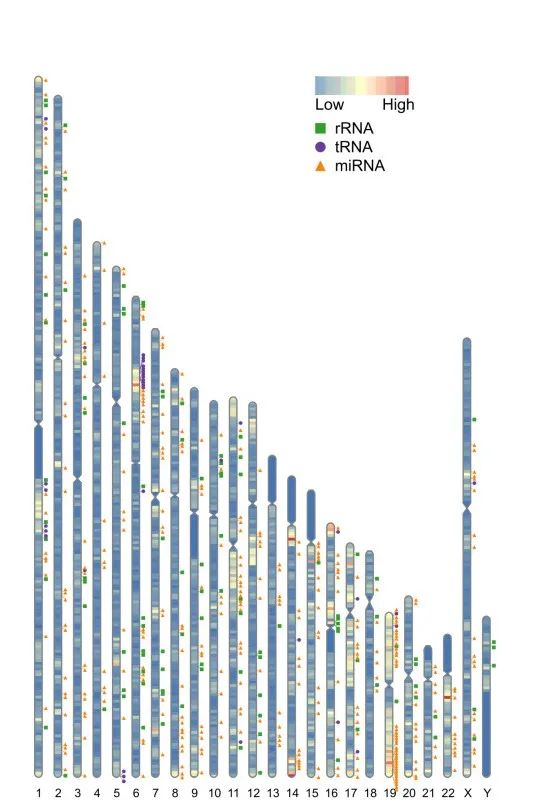
标签数据也可以用另一个热图来展示,与基因密度做对比:
ideogram(karyotype = human_karyotype, overlaid = gene_density, label = LTR_density, label_type = "heatmap", colorset1 = c("#f7f7f7", "#e34a33"), colorset2 = c("#f7f7f7", "#2c7fb8"),output = 'chromosome_10.svg') #use the arguments 'colorset1' and 'colorset2' to set the colors for gene and LTR heatmaps, separately.
convertSVG("chromosome_10.svg", device = "png",file = 'chromosome_10.png')## Warning in checkValidSVG(doc, warn = warn): This picture may not have been
## generated by Cairo graphics; errors may result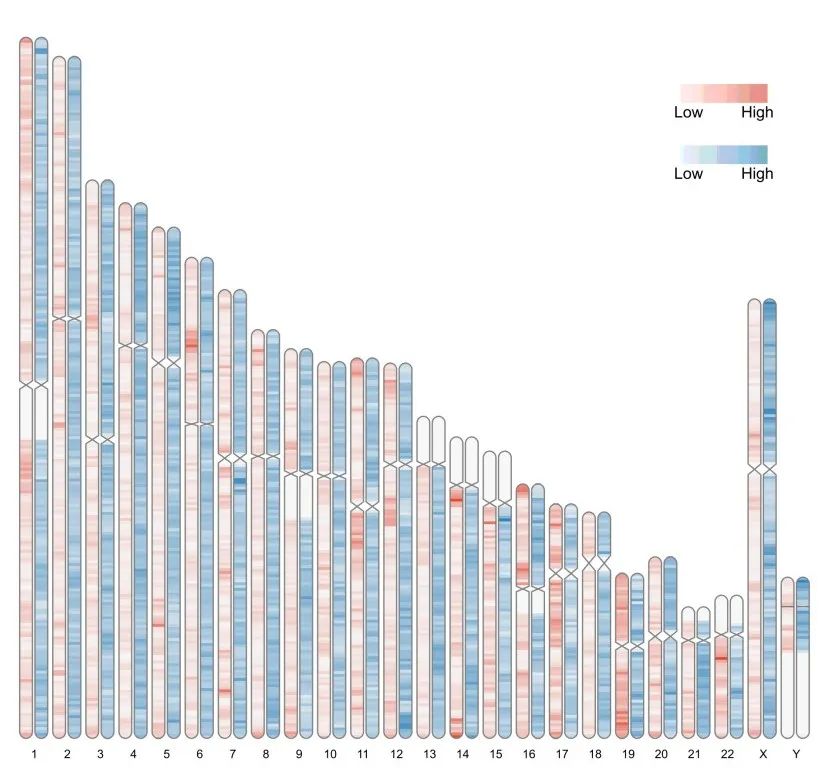
标签数据也可以做折线图展示:
# 这里我们加载一个新的测试数据:
data(liriodendron_karyotype, package="RIdeogram") # karyotype data,这个数据集包含了Liriodendron,即黄连木属,的染色体信息,
head(liriodendron_karyotype)## Chr Start End
## 1 1 0 118073833
## 2 2 0 98364873
## 3 3 0 207093695
## 4 4 0 50051714
## 5 5 0 45443526
## 6 6 0 35772468data(Fst_between_CE_and_CW, package="RIdeogram") #用于绘制热图的数据,这里是Fixation Index信息
head(Fst_between_CE_and_CW)## Chr Start End Value
## 1 1 1 2000000 0.0646357
## 2 1 1000001 3000000 0.0626714
## 3 1 2000001 4000000 0.0582397
## 4 1 3000001 5000000 0.0679570
## 5 1 4000001 6000000 0.0965196
## 6 1 5000001 7000000 0.0934111data(Pi_for_CE, package="RIdeogram") #用于绘制折线图的数据,这里是同染色体区域内的核苷酸多样性
head(Pi_for_CE)## Chr Start End Value Color
## 1 1 1 2000000 0.00273566 fc8d62
## 2 1 1000001 3000000 0.00239580 fc8d62
## 3 1 2000001 4000000 0.00319407 fc8d62
## 4 1 3000001 5000000 0.00286900 fc8d62
## 5 1 4000001 6000000 0.00186596 fc8d62
## 6 1 5000001 7000000 0.00186182 fc8d62绘制折线图标签:
ideogram(karyotype = liriodendron_karyotype, overlaid = Fst_between_CE_and_CW, label = Pi_for_CE, label_type = "line", colorset1 = c("#e5f5f9", "#99d8c9", "#2ca25f"),output = 'chromosome_11.svg')
convertSVG("chromosome_11.svg", device = "png",file = 'chromosome_11.png')## Warning in checkValidSVG(doc, warn = warn): This picture may not have been
## generated by Cairo graphics; errors may result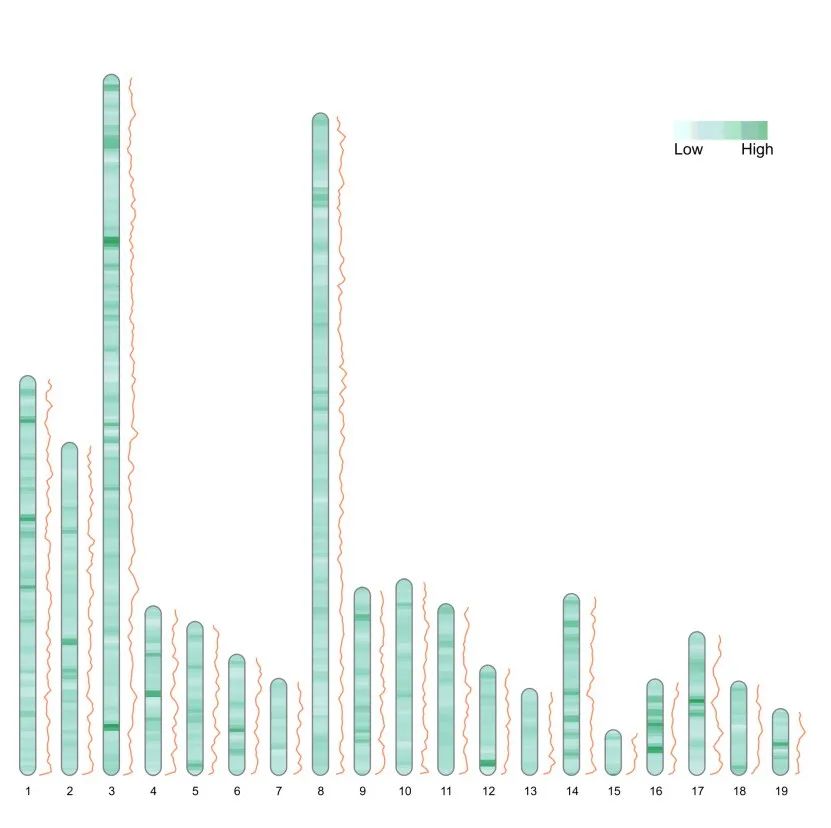
甚至可以绘制两条折线图:
data(Pi_for_CE_and_CW, package="RIdeogram")
ideogram(karyotype = liriodendron_karyotype, overlaid = Fst_between_CE_and_CW, label = Pi_for_CE_and_CW, label_type = "line", colorset1 = c("#e5f5f9", "#99d8c9", "#2ca25f"),output = 'chromosome_12.svg')
convertSVG("chromosome_12.svg", device = "png",file = 'chromosome_12.png')## Warning in checkValidSVG(doc, warn = warn): This picture may not have been
## generated by Cairo graphics; errors may result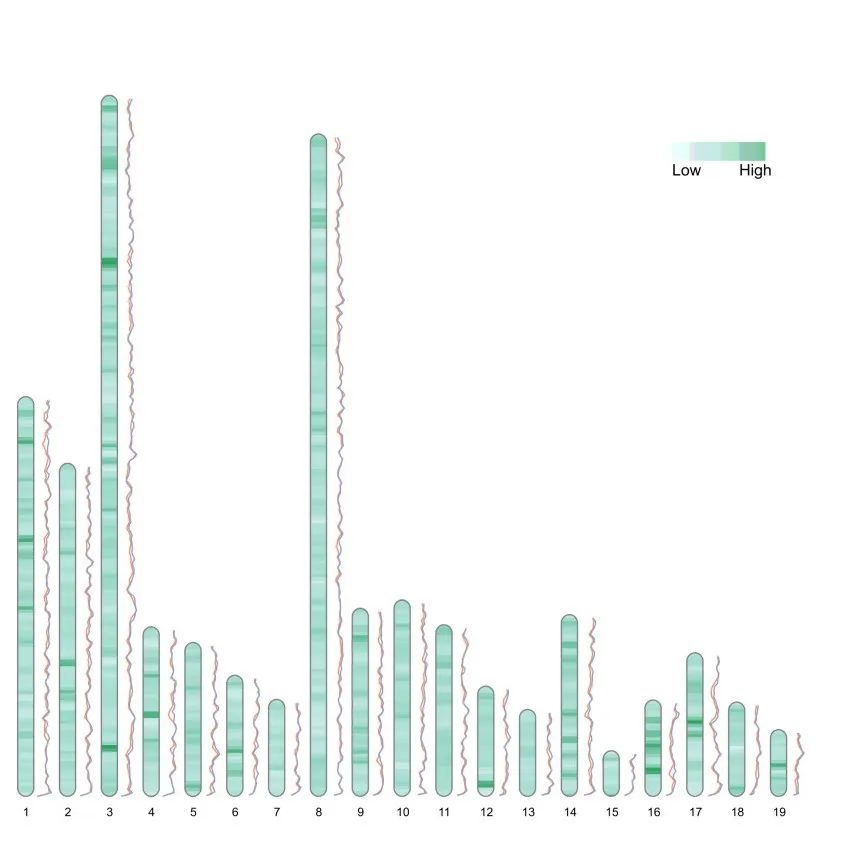
折线图也可以有填充:
ideogram(karyotype = liriodendron_karyotype, overlaid = Fst_between_CE_and_CW, label = Pi_for_CE_and_CW,
label_type = "polygon", # 设置多边形为填充方式
colorset1 = c("#e5f5f9", "#99d8c9", "#2ca25f"),output = 'chromosome_13.svg')
convertSVG("chromosome_13.svg", device = "png",file = 'chromosome_13.png')## Warning in checkValidSVG(doc, warn = warn): This picture may not have been
## generated by Cairo graphics; errors may result
三、多基因组对比
3.1 桑基图(用于两两比较)
### 需要用到新的数据:###
# 染色体背景数据:
data(karyotype_dual_comparison, package="RIdeogram")
head(karyotype_dual_comparison)# 第一列为染色体编号,第二、三列为起止位点,第四列是染色体填充颜色值,第五列是物种,第六列为物种label的大小,第七列为物种label的颜色## Chr Start End fill species size color
## 1 I 1 23037639 969696 Grape 12 252525
## 2 II 1 18779884 969696 Grape 12 252525
## 3 III 1 19341862 969696 Grape 12 252525
## 4 IV 1 23867706 969696 Grape 12 252525
## 5 V 1 25021643 969696 Grape 12 252525
## 6 VI 1 21508407 0ab276 Grape 12 252525# 这个数据中包含"Grape" "Populus"两个物种的
table(karyotype_dual_comparison$species)##
## Grape Populus
## 19 19# 对比数据:
data(synteny_dual_comparison, package="RIdeogram")
head(synteny_dual_comparison)# 第一列为物种1的区域编号,第二、三列为该区域的起止位点,四~六列同理,第七列为桑基图连线及对应区域填充颜色## Species_1 Start_1 End_1 Species_2 Start_2 End_2 fill
## 1 1 12226377 12267836 2 5900307 5827251 cccccc
## 2 15 5635667 5667377 17 4459512 4393226 cccccc
## 3 9 7916366 7945659 3 8618518 8486865 cccccc
## 4 2 8214553 8242202 18 5964233 6027199 cccccc
## 5 13 2330522 2356593 14 6224069 6138821 cccccc
## 6 11 10861038 10886821 10 8099058 8011502 cccccc# 绘制起来很简单:
ideogram(karyotype = karyotype_dual_comparison, synteny = synteny_dual_comparison,output = 'sankey_chromosome.svg')
convertSVG("sankey_chromosome.svg", device = "png",file = 'sankey_chromosome.png')## Warning in checkValidSVG(doc, warn = warn): This picture may not have been
## generated by Cairo graphics; errors may result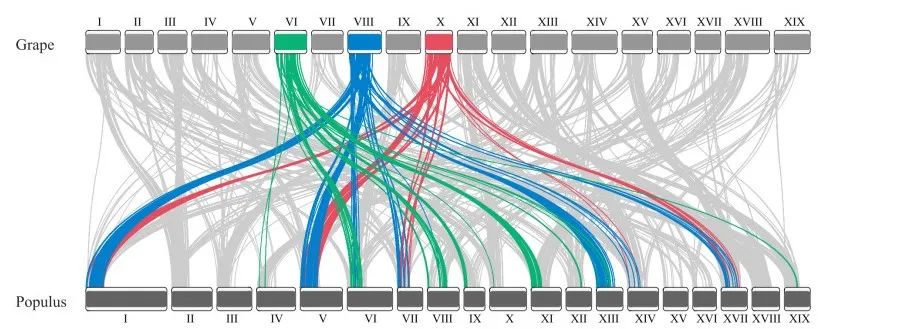
## 3.2 三元比较
数据与桑基图部分类似,只不过变成了三个物种的数据:
data(karyotype_ternary_comparison, package="RIdeogram")
head(karyotype_ternary_comparison)## Chr Start End fill species size color
## 1 NA 1 15980527 fcb06b Amborella 10 fcb06b
## 2 NA 1 11522362 fcb06b Amborella 10 fcb06b
## 3 NA 1 11085951 fcb06b Amborella 10 fcb06b
## 4 NA 1 10537363 fcb06b Amborella 10 fcb06b
## 5 NA 1 9585472 fcb06b Amborella 10 fcb06b
## 6 NA 1 9414115 fcb06b Amborella 10 fcb06btable(karyotype_ternary_comparison$species)##
## Amborella Grape Liriodendron
## 100 19 19data(synteny_ternary_comparison, package="RIdeogram")
head(synteny_ternary_comparison)## Species_1 Start_2 End_2 Species_2 Start_1 End_1 fill type
## 1 1 4761181 2609697 1 342802 981451 cccccc 1
## 2 6 6344197 8074393 1 15387184 16716190 cccccc 1
## 3 10 6457890 9052487 1 11224953 14959548 cccccc 1
## 4 13 6318795 1295413 1 20564870 21386271 cccccc 1
## 5 16 1398101 2884119 1 21108654 22221088 cccccc 1
## 6 16 1482529 2093625 1 21864494 22364888 cccccc 1tail(synteny_ternary_comparison, n = 20)## Species_1 Start_2 End_2 Species_2 Start_1 End_1 fill type
## 571 16 19278042 20828694 2 95267449 93334736 cccccc 3
## 572 12 20546006 22461088 2 22647943 18365764 cccccc 3
## 573 4 22259262 23453956 2 15068249 17839485 cccccc 3
## 574 14 22377895 23821929 2 97299880 96033346 cccccc 3
## 575 6 1538773 2808373 1 91285578 95681546 cccccc 3
## 576 11 3381792 4954528 1 67689752 75286468 cccccc 3
## 577 9 4814481 6975840 1 69506847 76015710 cccccc 3
## 578 10 7091825 9742616 1 19333526 24516133 cccccc 3
## 579 13 22063957 23402389 1 95843870 92195256 cccccc 3
## 580 7 679765 1881756 6 7365421 7531534 e41a1c 1
## 581 7 679765 2752867 13 501561 766473 e41a1c 1
## 582 7 679765 3012501 8 7406703 8222490 e41a1c 1
## 583 7 2049369 2942034 14 29350547 34369929 e41a1c 2
## 584 7 2075095 1538540 10 28985737 30815217 e41a1c 2
## 585 13 531939 834472 14 28866243 35278211 e41a1c 3
## 586 8 7427221 8894821 14 28632063 34805893 e41a1c 3
## 587 6 7567597 7690342 14 32050301 34913801 e41a1c 3
## 588 13 501561 876423 10 30496700 27874100 e41a1c 3
## 589 6 7171014 7815454 10 31408837 27660041 e41a1c 3
## 590 8 5773528 9346871 10 31408837 26585934 e41a1c 3可视化依然很简单:
ideogram(karyotype = karyotype_ternary_comparison, synteny = synteny_ternary_comparison,output = 'tri_chromosome.svg')
convertSVG("tri_chromosome.svg", device = "png",file = 'tri_chromosome.png')## Warning in checkValidSVG(doc, warn = warn): This picture may not have been
## generated by Cairo graphics; errors may result
连线的颜色可以是渐变的:
data(synteny_ternary_comparison_graident, package="RIdeogram")
ideogram(karyotype = karyotype_ternary_comparison, synteny = synteny_ternary_comparison_graident, output = 'tri_chromosome_graident.svg')
convertSVG("tri_chromosome_graident.svg", device = "png",file = 'tri_chromosome_graident.png')## Warning in checkValidSVG(doc, warn = warn): This picture may not have been
## generated by Cairo graphics; errors may result
最后,保存所有数据的镜像备用:
save.image('biomamba.rdata')
















 1887
1887

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









