






生信碱移
贝叶斯NMF算法
病理、生理相关的生物学过程中,不同基因各司其职。一个普遍的假设是,执行特定功能的生物学过程往往具有一定的表达模式。非负矩阵分解(NMF)是一种适合高通量生物学的无监督聚类方法。然而,仅仅从 NMF 结果推断生物过程还是需要额外的事后统计和注释分析,否则无法很好的解释学习到的基因特征规律。
为此,来自美国约翰霍普金斯大学的研究者于 2023年12月11日在Nature Protocols [IF:13.1] 杂志分享了一套流程,其中介绍了贝叶斯NMF算法(CoGAPS),并演示了如何通过 CoGAPS 分析量化公共领域单细胞 RNA 测序数据中的细胞状态转换。

▲ 10.1038/s41596-023-00892-x
最近看到CoGAPS包好像发了几篇高分文章,便赶紧给各位铁子介绍一下如何 R 包CoGAPS执行非负矩阵分解算法CoGAPS。上面的文章也同时提供了python的版本,大家感兴趣的话也可以自行学习:
-
https://github.com/FertigLab/pycogaps
-
https://github.com/FertigLab/CoGAPS/
1 R包的安装与引用
可以使用以下代码安装与引用该R包:
#devtools::install_github("FertigLab/CoGAPS")
library(CoGAPS)
library(Seurat)
library(dplyr)
library(viridis)
2 数据的读入
示例数据inputdata.Rds可以在以下链接下载:
-
https://github.com/FertigLab/CoGAPS/blob/b6b849cf84ed91f34038048e2b29ea0fcf93570b/data/inputdata.Rds
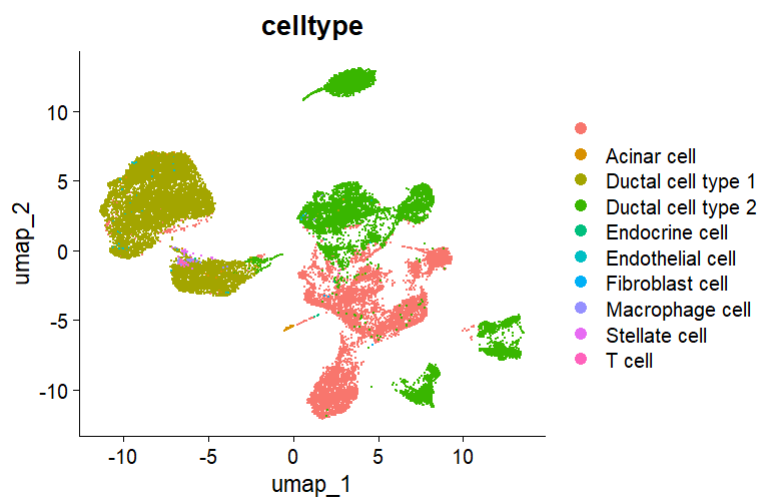
这个inputdata.Rds其实就是我们的 seurat 对象。这里我们可以将其读入并命名为af,看看初步的细胞分群:
af <- readRDS("inputdata.Rds")
DimPlot(af, pt.size = 0.2, group.by = "celltype", reduction = "umap")
3 执行 CoGAPS 分析
① 提取count矩阵进行CoGAPS分析,注意nPatterns参数,下方设置为3,即指定了3种表达模式(英文: pattern):
# 提取count矩阵
pdac_epi_counts <- GetAssayData(object = af, slot = "counts") %>% as.matrix()
norm_pdac_epi_counts <- log1p(pdac_epi_counts)
# 参数设置
pdac_params <- CogapsParams(nIterations=50000, # 迭代数量
seed=42, # 设置随机数种子
nPatterns=3, # 设置聚类数量
sparseOptimization=TRUE, # optimize for sparse data
distributed="genome-wide") # parallelize across sets
# 正式执行分析
pdac_epi_result <- CoGAPS(norm_pdac_epi_counts, pdac_params)
saveRDS(pdac_epi_result, "./pdac_epi_cogaps_result.rds")
② 可视化细胞的表达模式评分:
# 可视化细胞的表达模式评分
cogapsresult <- readRDS("./pdac_epi_cogaps_result.rds")
patterns_in_order <-t(cogapsresult@sampleFactors[colnames(af),])
af[["CoGAPS"]] <- CreateAssayObject(counts = patterns_in_order)
DefaultAssay(af) <- "CoGAPS"
pattern_names <- rownames(af@assays$CoGAPS)
color_palette <- viridis(n=10)
FeaturePlot(af, pattern_names, cols=color_palette, reduction = "umap") & NoLegend()
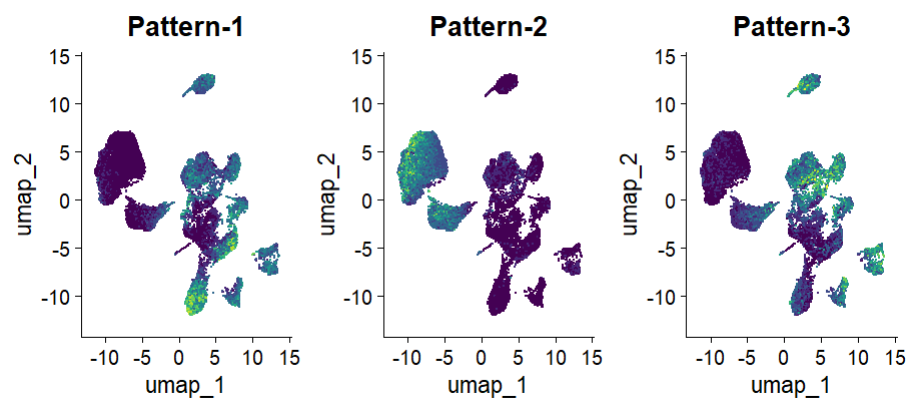
简单来讲,执行COGA分析后,每个细胞对于每个表达模式 pattern 都会获得一个相应的评分。还可以使用常规的小提琴图展示或者自行进行更多的探索:
# 进行gsea分析
VlnPlot(af, features = pattern_names, group.by = "celltype", pt.size = 0.2)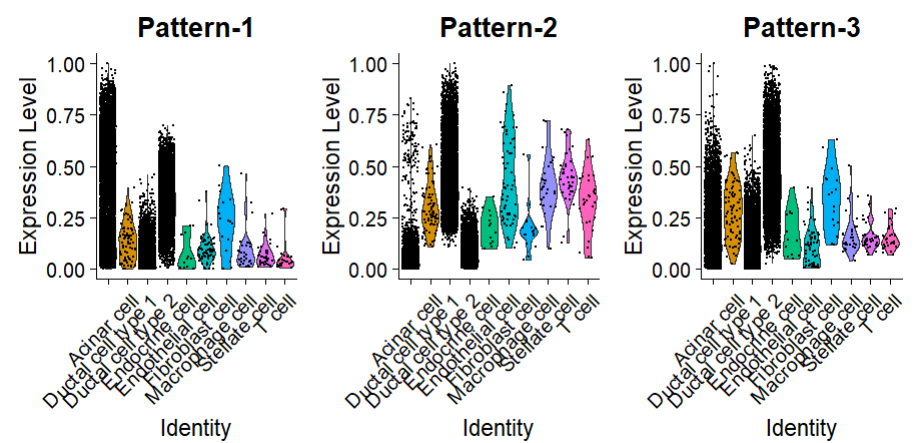
③ 计算每个表达模式的标志基因,下方代码运行后将在genes_pm储存中每个基因在不同模式中的排名:
pm <- patternMarkers(cogapsresult, threshold="cut")
genes_pm <- pm$PatternRanks
genes_pm[1:5, 1:3]
# Pattern_1 Pattern_2 Pattern_3
#SAMD11 1 11496 724
#NOC2L 10980 11147 10890
#KLHL17 1128 100 11802
#PLEKHN1 86 13506 10504
#HES4 8083 1572 13497
④ 执行 GSEA 富集分析,通过指定plotPatternGeneSet函数中的whichpattern参数可以指定表达模式。
# 进行gsea分析
geneset <- read.gmt("h.all.v7.5.1.symbols.gmt") # 读取基因集,这里使用的是msigdb数据库中的hallmark基因集,也可以自定义
geneset <- split(geneset$gene, geneset$term)
hallmarks <- getPatternGeneSet(object = cogapsresult, gene.sets = geneset, method = "enrichment")
plotPatternGeneSet(hallmarks, whichpattern = 1, padj_threshold = 0.05)
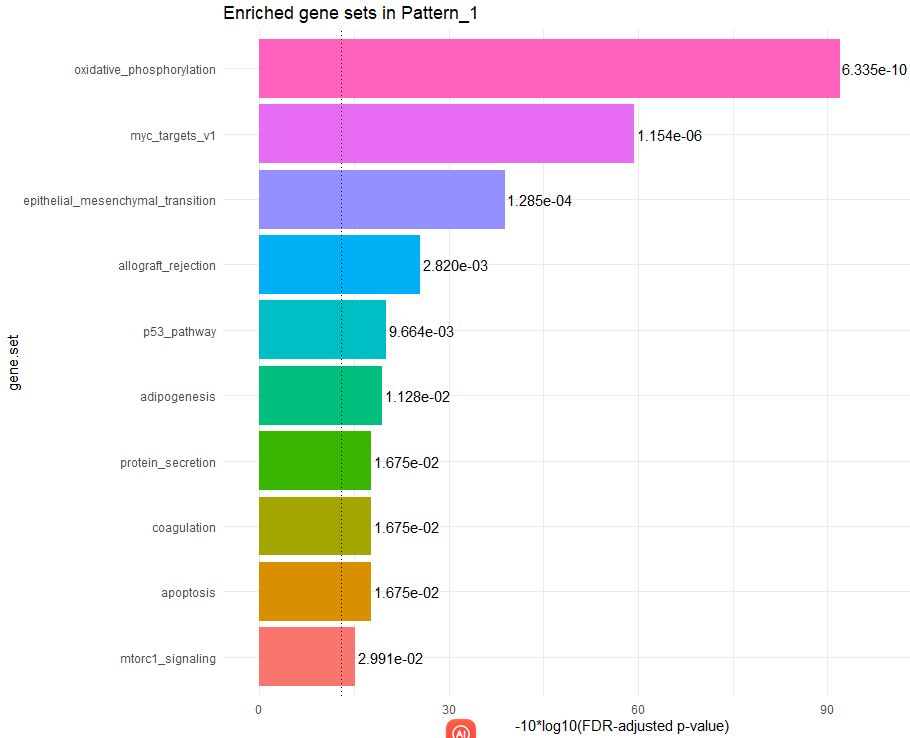
注意,getPatternGeneSet函数的method参数支持两种设置enrichment与overrepresentation,下面是它们各自的介绍:
-
enrichment or overrepresentation. Conducts a test for gene set enrichment using fgsea::gsea ranking features by pattern amplitude or a test for gene set overrepresentation in pattern markers using fgsea::fora, respectively.
此外,getPatternGeneSet函数还需要提供富集使用基因集列表,可以自行准备或下载。上面的示例代码读入了 msigdb 数据库的 hallmark 基因集(gmt文件格式),下载链接如下:
-
https://www.gsea-msigdb.org/gsea/msigdb/collections.jsp
今天就分享到这
欢迎点击关注

















 182
182

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









