






生信碱移
68种单细胞批次整合方法的比较
整合多个批次的单细胞数据往往是把研究做大的第一步,来自德国慕尼黑工业大学的研究者测试了已有的 68 种单细胞批次整合方法,应该认为各位老铁提供一些选择参考。
单细胞图谱通常包括跨越不同位置、实验室和条件的样本,导致数据中存在复杂的嵌套批次效应。目前,已经发表的文献中存在多种单细胞批次整合方法,并没有一致的评估标准来用于选择最合适的方法。来自,来自德国慕尼黑工业大学的研究者使用了85 个批次的基因表达、染色质可及性和模拟数据,涵盖 120 万细胞与 13 个图谱整合任务,测试了已有的 68 种单细胞批次整合方法。作者根据可扩展性、易用性和其在去除批次效应同时保留生物变异性方面的表现来评估不同的整合方法与预处理组合(涉及 14 个评估指标)。
▲ DOI: 10.1038/s41592-021-01336-8
研究结果表明,高变基因的选择可以提高数据整合方法的性能,而数据缩放则促使方法优先考虑去除批次效应而非保留生物变异性。总体而言,scANVI、Scanorama、scVI 和 scGen 表现良好,尤其是在复杂整合任务中表现尤为突出,而单细胞 ATAC-测序的整合性能则强烈依赖于特征空间的选择。作者还提供了免费 Python 模块和基准测试管道用于识别新数据的最佳整合方法, 用于 benchmark 新方法并改进已有方法:
-
https://github.com/theislab/scib-reproducibility
一、基于 16 种方法、13 项任务与 14 个指标的单细胞数据整合基准测试简介
该研究通过搭建 单细胞整合评估平台(scIB),在 13 个整合任务(含 2 个模拟任务、5 个 scRNA-seq 任务和 6 个 scATAC-seq 任务)上。为检验方法的准确性、可用性和可扩展性,研究设计了 14 个性能指标从批次效应移除与生物学变异保持两大维度进行量化评估;同时,考虑了多种输出格式(嵌入、矫正矩阵、集成图等)及不同的预处理策略(含/不含缩放与高变基因筛选)。最终对高达 68 种不同方法与预处理组合进行了 590 次整合尝试,并通过新开发的指标(如基于图的 kBET 和 LISI 等)对结果进行多方面衡量,力求为用户提供可重复、可拓展的单细胞整合方法评估与选择框架。
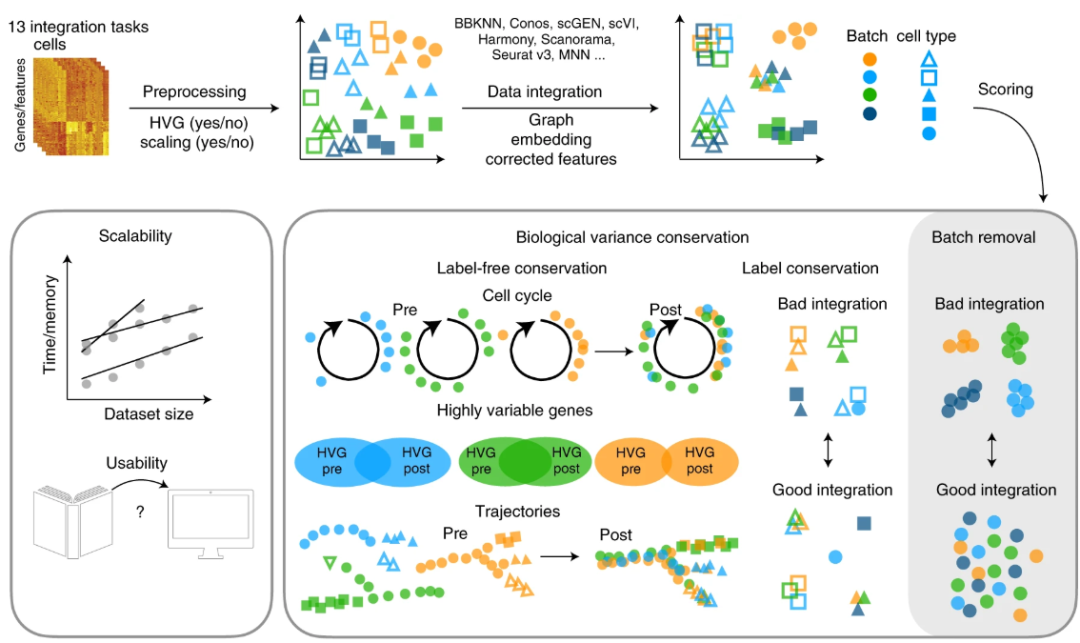
▲ 研究整体设计与评估方法。
二、在人类免疫细胞整合中的评估:多种方法在批次效应移除与生物学保真间的表现
在人类免疫细胞整合任务中,研究团队针对来自五个数据集、共计十个不同批次(供体)的外周血和骨髓细胞进行了整合测试。结果显示,Scanorama (embedding 方式)、FastMNN (embedding 方式)、scANVI 以及 Harmony 的综合表现最优:它们在大幅减小批次间差异的同时,较好地保留了细胞类型及亚型的生物学特征。具体来看,Scanorama 对 Smart-seq2 与 10X 等不同测序平台的整合效果更出色,scANVI 则在处理完整转录本(Smart-seq2)数据时出现剩余批次结构,Harmony 在维持孤立标签的生物保真度上表现不足。对于红细胞发育轨迹的还原,表现优异的方法均能保留从造血干细胞到成熟红细胞的连续性;而 DESC、scGen 和 Seurat v3 CCA 等方法则分别因过度聚类、缺失生物学潜在结构或过度合并细胞类型等原因导致轨迹保留不佳。
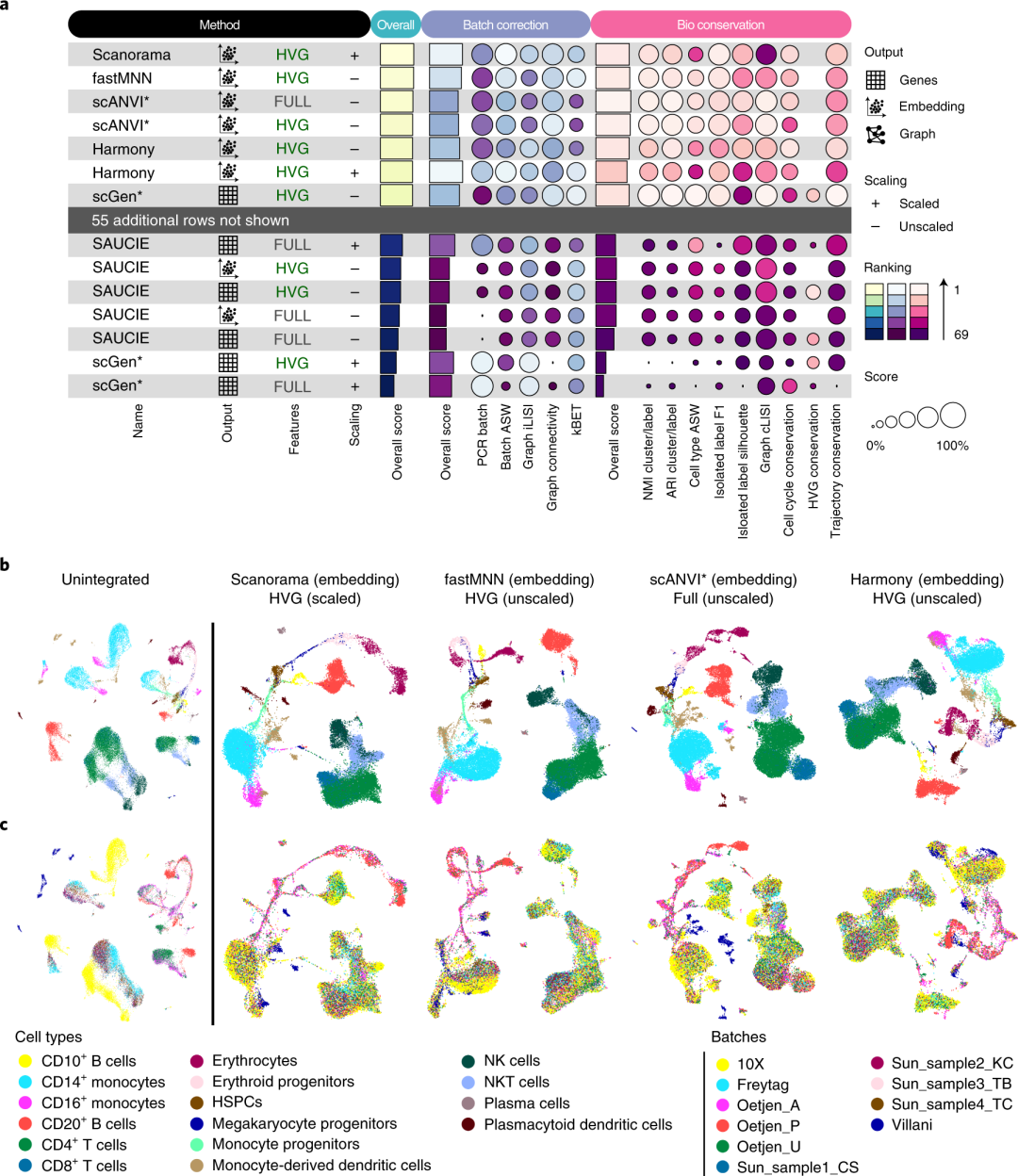
▲ 多种方法在免疫细胞整合测试中的排名,图a只展示了部分结果,所有排名可以在补充文件获得。
三、多种场景与不同预处理策略的单细胞整合评估
在对五个 scRNA-seq 实际数据集和两个模拟数据集的整合任务进行评估后发现,整合方法的表现在不同任务间存在差异,主要受生物学复杂度和批次差异强度影响。通常,越复杂的生物学场景越容易出现“批次效应移除”与“生物学变异保持”之间的权衡。例如,Seurat v3、Harmony 等在较简单的数据集或模拟情形下表现优异,但在更复杂的真实数据中可能无法兼顾这两方面;而 Scanorama、scVI 及具备细胞类型先验信息的 scANVI 能在保留更多生物学信息的同时,仍然显著减少批次效应。此外,scGen 与 scANVI 在生物学特征编码越精细(如细胞类型分辨更高)时,其整合效果越好,特别是在跨物种或跨测序模式(单细胞 vs. 单核)整合时能更好地平衡差异与变异。
在预处理方面,高变基因(HVG)筛选多半能带来更好的综合得分;而是否进行 scaling(标准化处理)则因方法而异,总体上有利于强化批次效应移除却会牺牲生物学变异保持。基于邻近匹配(MNN)或深度学习(如 scVI、trVAE)的方法在细胞数目庞大、批次结构复杂时往往表现更佳。若使用 CPU,scGen 在超大规模数据(如百万级小鼠脑细胞数据)上的运行效率可能受到限制。综合排名显示,Scanorama(embedding 输出)、scVI 及 scANVI 在更真实的复杂场景中表现最为出色;而 SAUCIE、DESC 等方法得分较低。对需要跨物种整合且注重全球连续发育轨迹的应用而言,仍面临较大挑战,需要在保留精细生物学差异与减少批次效应之间进行权衡与选择。
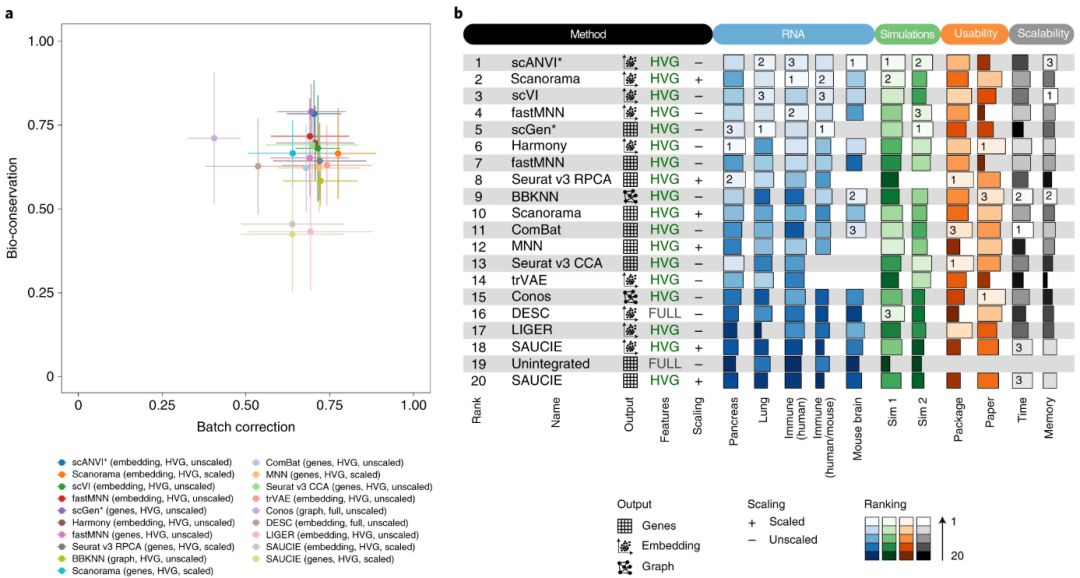
▲ 所有 RNA 整合任务和模拟的基准测试结果概述,包括易用性和可扩展性。
四、单细胞 ATAC-seq 批次整合:特征空间选择、可扩展性与方法平衡性评估
在三套小鼠大脑单细胞 ATAC-seq 数据(不同技术平台)上进行整合测试,结果显示不同特征空间(峰、窗口及基因活性)对整合效果影响明显:仅有不到三成(27%)的整合结果能优于未整合状态(尤其在峰特征空间中表现相对更好,基因活性特征空间则整体生物变异度较低)。在 ATAC 任务中,多数方法的批次效应移除能力和生物学保真难以兼顾:
-
LIGER 在峰和窗口两个特征空间中表现相对稳定,能较好地去除批次差异,但可能引入新的亚群结构;
-
Harmony 整合后保留了较多生物学差异,但仍会出现部分小型神经元亚群的重叠;
-
Seurat v3 RPCA、BBKNN 在小规模 ATAC 整合中具备一定平衡性,却无法彻底去除所有批次效应。
-
在可扩展性评估中,由于 ATAC-seq 特征数量庞大,仅 7/16 的方法能运行在含 9.4 万以上特征的“大规模”任务上。ComBat、BBKNN 和 SAUCIE 的运行时间最短,而 scVI、scANVI 与 BBKNN 最节省内存;相较之下,MNN、scGen 和 trVAE 对大规模数据的 CPU 时间或内存需求不具可行性。另外,在可用性(文档、教程、维护度及鲁棒性验证)上,Harmony、Seurat v3 与 BBKNN 表现最好,而 DESC、scANVI 及 trVAE 仍需改进。
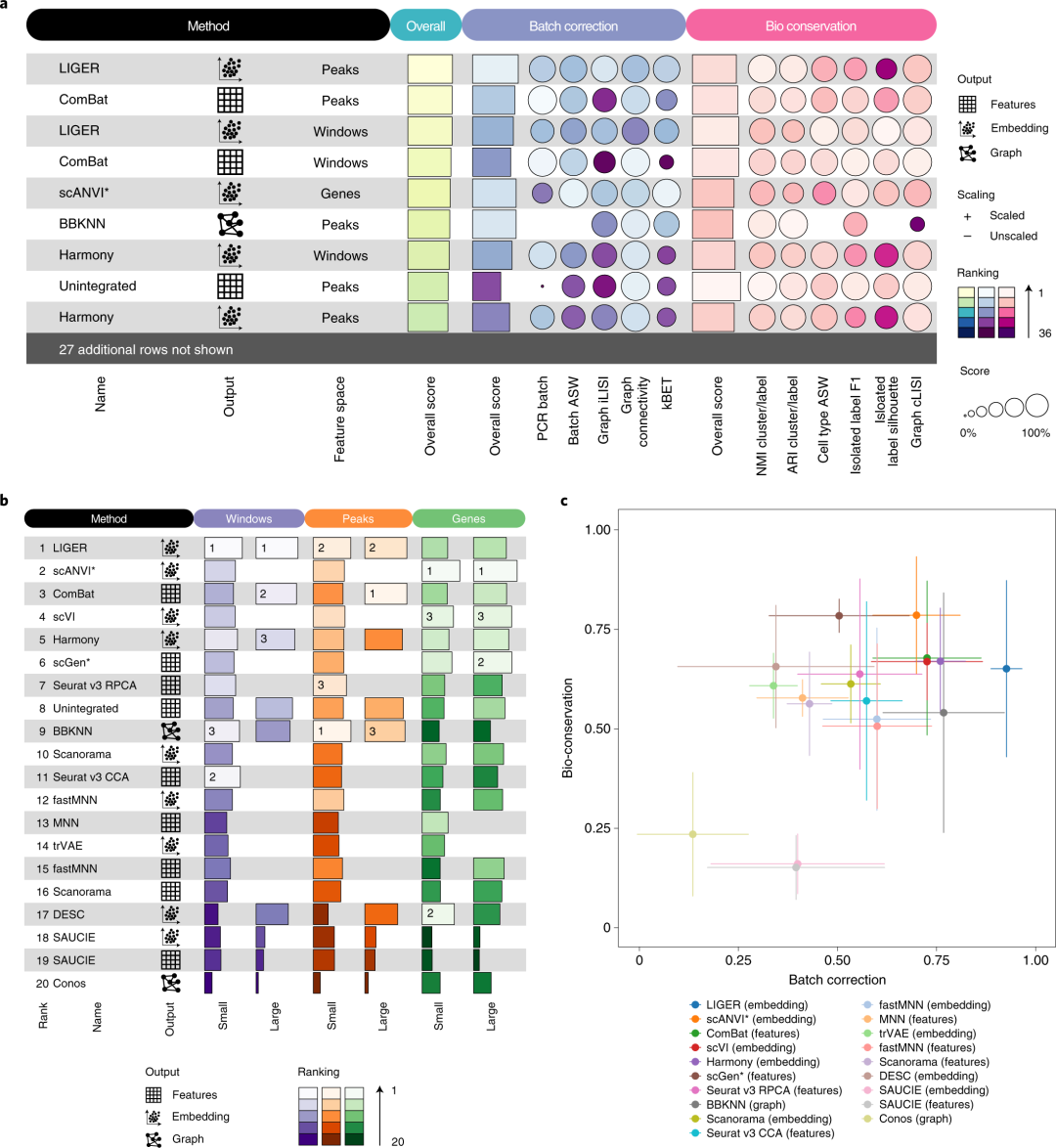
▲ 鼠脑 ATAC 整合任务的基准测试结果。
总结
在本基准研究中,作者使用 14 项指标对 16 种整合方法与 4 种预处理方式进行了综合评估,涵盖 13 个具备不同复杂度的单细胞整合任务(包括五个 scRNA-seq、两个模拟以及多个 scATAC-seq 任务)。结果发现,方法表现与数据本身的生物学复杂度和批次差异强弱紧密相关:
-
Harmony 适合较简单且批次信号明显的数据,但在真实且复杂的场景中通常无法排入前三(这个排名已经超乎小编的预期了,没想到Harmony的排名在60多种方法里面仍然具备一定竞争力);当细胞类型注释可用时。
-
scGen 和 scANVI 能出色地平衡批次校正与生物学变异保留;若缺乏注释或数据量足够大,scVI 与 Scanorama 同样在复杂任务中表现优异;
-
对于 scATAC-seq 数据,LIGER 与 Harmony 侧重去除强批次效应,但可能牺牲某些生物学细节;以峰或窗口为特征空间通常能更好保持细胞类型特征;
-
深度学习方法(scGen、scANVI、scVI、trVAE 等)在更大规模、批次多样的任务上具备潜力,但对计算资源(如 GPU)和超参数优化的需求更高;
-
就可扩展性与实用性而言,BBKNN、ComBat、SAUCIE 在运行时间上表现相对较好,scVI、scANVI 在内存消耗上更具优势,而部分方法(如 MNN、scGen、trVAE)在百万级或上十万特征的整合任务上表现不佳。
作者最后给出了一套基于可用性、可扩展性与预期表现的选型建议,并指出如需兼顾罕见细胞状态发现与批次校正,应谨慎评估是否需要去除某些“混合”了生物信号的批次差异。此外,结合自己的数据规模、下游分析需求(如轨迹分析、基因模块挖掘等)以及对 GPU 等硬件资源的可用程度,选择合适的整合方法至关重要。

▲ 作者最后提供的图片,可以用于指导集成方法的选择。
今天就分享到这


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









