






生信碱移
空转+GWAS
gsMap将空间转录组学与全基因组关联分析结合起来,以空间分辨的方式将空间点映射到人类复杂性状(包括疾病)。
组织中细胞的组成与空间结构对其功能至关重要,也可作为健康/疾病状况的指标。例如,肝小叶中肝细胞的放射状空间排列有助于高效代谢血液中的营养物质,而大脑中神经元的空间分布与连接网络则调控情绪、认知和行为。空间转录组学(ST)技术的进步使得在细胞原位空间位置解析基因表达成为可能,其与单细胞测序技术相比提供了更多维度的生物学信息。
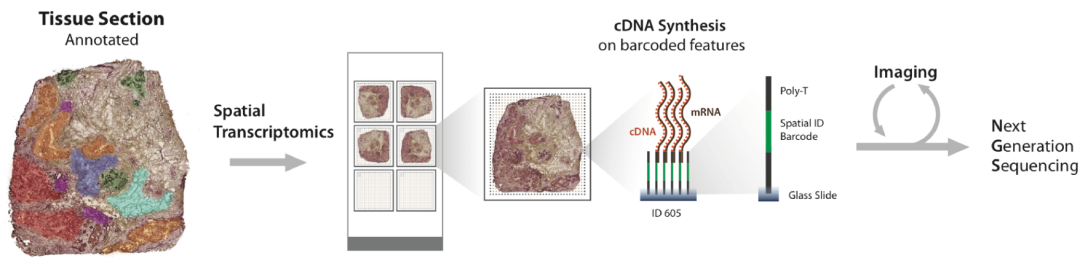
▲ 一、空间转录组测序技术原理:空间转录组学是一种可以对组织切片中的转录本库进行定量分析的方法。通过将组织切片放在载玻片上,载玻片上排列有包含记录位置信息的寡核苷酸条形码,后者可以生成具有精确位置信息的高质量 cDNA 文库,用于 RNA 测序。
为鉴定性状相关细胞或类型,先前研究提出遗传学驱动的策略,将复杂性状的全基因组关联分析(GWAS)数据与单细胞RNA测序(scRNA-seq)数据整合。其核心原理在于评估性状的遗传关联信号是否富集于特定细胞群高表达基因的邻近区域。尽管这些方法可识别性状相关细胞,但由于scRNA-seq缺乏空间信息,其难以定位细胞的空间分布。理论上,此类方法可应用于ST数据,但由于未纳入细胞空间信息且ST数据噪声较高,其在空间分辨的性状相关细胞定位中效力有限。
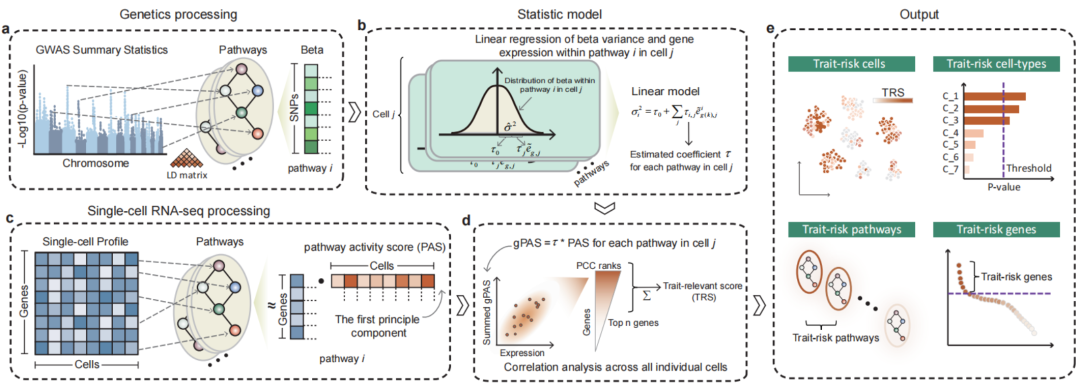
▲ 二、scPagwas算法原理:基于GWAS与单细胞进行性状关联细胞鉴定。具体来讲,scPagwas 采用多基因回归模型来优先考虑一组与性状相关的基因,并通过结合通路活性转化的单细胞 RNA 测序(scRNA-seq)数据与全基因组关联研究(GWAS)摘要数据,揭示与性状相关的细胞亚群。DOI:10.1016/j.xgen.2023.100383。
为此,来自西湖大学的杨剑团队提出遗传学驱动的复杂性状空间细胞定位方法gsMap,通过整合高分辨率ST数据与GWAS汇总统计数据,实现性状相关细胞的空间解析定位。研究团队利用覆盖25个器官的胚胎ST数据集,通过模拟实验验证了gsMap的性能,并通过复现已知的性状相关细胞或区域证实了其敏感性。值得注意的是,杨剑团队同时也开发了SMR软件,所以这个工具还是有很大的学习价值的(尽管现在还处于预印本阶段)。
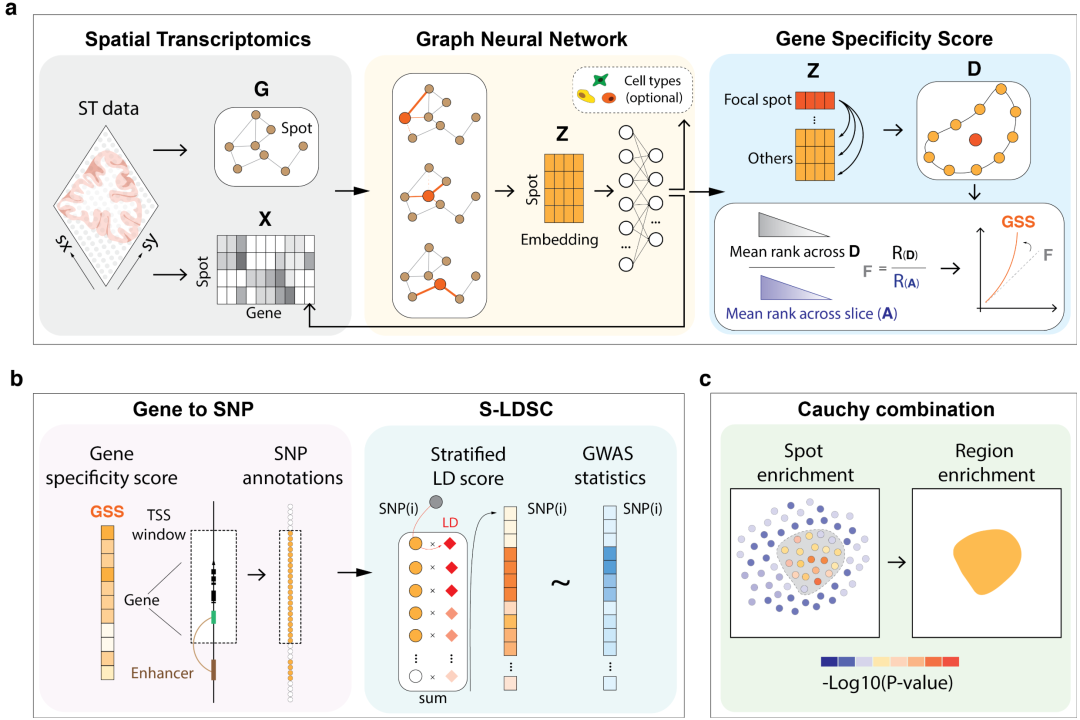
▲ 三、gsMap方法框架:a.通过GNN整合基因表达、空间坐标与细胞类型先验信息,基于嵌入向量余弦相似性识别目标位点的同质位点(微域),通过对比基因在微域内与全局ST数据的表达排序计算位点特异性评分;b.将位点GSS映射至SNP,利用S-LDSC评估SNP-GSS注释的性状遗传力富集;c.通过柯西组合检验整合空间区域内的位点P值。
gsMap包含三步流程:①基于spot同质性降低ST数据的稀疏性与技术噪声,利用图神经网络(GNN)识别每个目标spot的同质spot(即基因表达模式与空间位置相似的测序点),通过整合同质spot信息计算各基因在目标spot的基因特异性评分(GSS),用于反映基因在特定位点的相对表达水平(上图a);②将位点的GSS基于基因转录起始位点(TSS)距离及表观基因组数据建立的SNP-基因关联图谱映射至SNP,生成每位个点独特的SNP-GSS注释。随后基于分层连锁不平衡评分回归(S-LDSC),评估高GSS的SNP是否显著富集目标性状的遗传力,并基于富集P值量化位点与性状的关联显著性(上图b);③采用柯西组合检验整合空间区域内所有位点的P值,量化特定区域与性状的关联显著性(上图c)。本质上讲,gsMap的核心原理是评估ST数据中某位点(spot)高表达基因邻近的遗传变异(SNP)是否富集于目标性状的遗传关联信号。
gsMap可以用于性状相关空间区域以及致病基因识别等分析。其Github路径以及官方教程如下,其可以通过快速或逐步两种运行方式使用,同时提供了一些个性化设置:
-
https://github.com/JianYang-Lab/gsMap
-
https://yanglab.westlake.edu.cn/gps_data/website_docs/html/index.html
本文简单介绍一下该工具的输入以及快速运行模式。
0.软件安装
①gsMap基于python解释器,可以通过pip进行安装:
# 对python版本有限制
conda create -n gsMap python>=3.10
conda activate gsMap
pip install gsMap
也可以使用源码安装:
git clone https://github.com/JianYang-Lab/gsMap
cd gsMap
pip install -e .
安装以后可以运行下方代码进行测试:
gsmap --version
01.输入数据示例
①输入ST数据需为 h5ad 格式文件,须包含以下数据槽:
-
基因表达矩阵:存储于
layers["count"]属性中 -
空间坐标:存储于
obsm["spatial"]属性中 -
位点标签-可选:细胞/位点类型标签可存储于
obs["annotation"]属性
import scanpy as sc
# 读取h5ad文件
adata = sc.read_h5ad("gsMap_example_data/ST/E16.5_E1S1.MOSTA.h5ad")
# 验证数据结构
print("基因表达矩阵维度:", adata.layers["count"].shape)
print("空间坐标维度:", adata.obsm["spatial"].shape)
print("前五位细胞类型分布:\n", adata.obs["annotation"].value_counts().head())
②输入的GWAS汇总统计数据需整理为指定标准格式.gz,至少需要包含以下列:
-
SNP:SNP rs编号 -
Z:Z统计量 -
N:样本量
通过以下代码可以将你的GWAS汇总数据转换为上述压缩的标准化格式:
# 下载示例GWAS数据并解压
wget https://portals.broadinstitute.org/collaboration/giant/images/4/4e/GIANT_HEIGHT_YENGO_2022_GWAS_SUMMARY_STATS_ALL.gz
gzip -d GIANT_HEIGHT_YENGO_2022_GWAS_SUMMARY_STATS_ALL.gz
# 将GWAS汇总统计数据转换为所需格式
gsmap format_sumstats \
--sumstats 'GIANT_HEIGHT_YENGO_2022_GWAS_SUMMARY_STATS_ALL' \
--out 'HEIGHT'
# HEIGHT.sumstats.gz 文件为输出,简单查看以下前面几列
zcat HEIGHT.sumstats.gz | head -n 5
SNP A1 A2 Z N
rs3131969 G A 0.328 1494218.000
rs3131967 C T 0.386 1488150.000
rs12562034 A G 1.714 1554976.000
rs4040617 G A -0.463 1602016.000
02.快速执行分析
gsmap的Quick Mode 选项提供了一种简化和高效的方式来执行整个 gsMap 流水线。它通过利用基于 1000Genomes 项目欧洲人群的参考面板(1000G EUR)和来自 Gencode v46 gtf 文件的蛋白编码基因预计算权重。此选项非常适合喜欢简化方法使用的用户。如果想定制你的分析,例如使用自定义 GTF 文件、参考面板和可调参数,可以自行学习教程中的逐步运行部分。
以下是该模式需要的预计算文件(可以使用下载的文件):
-
基因组注释文件(GTF格式)
-
LD参考面板(基于1000G_EUR_Phase3的SNP-基因关联矩阵)
-
SNP权重文件(校正SNP-性状关联统计量的相关性)
-
同源基因转换文件(跨物种基因名映射,可选)
# 下载提供的预计算文件
wget https://yanglab.westlake.edu.cn/data/gsMap/gsMap_resource.tar.gz
tar -xvzf gsMap_resource.tar.gz
# 目录结构如下:
gsMap_resource
├── genome_annotation # 基因组注释(增强子、GTF)
├── homologs # 同源基因映射文件(人类-小鼠/猕猴)
├── LD_Reference_Panel # LD参考面板(1000G_EUR)
├── LDSC_resource # LDSC分析资源
└── quick_mode # 快速模式预计算矩阵
除此之外,则是由用户自己准备的GWAS汇总数据与空转数据:
# 下载示例数据
wget https://yanglab.westlake.edu.cn/data/gsMap/gsMap_example_data.tar.gz
tar -xvzf gsMap_example_data.tar.gz
# 目录结构如下:
gsMap_example_data/
├── GWAS # GWAS汇总统计文件
└── ST # 小鼠胚胎空间转录组数据(h5ad格式)
正式执行分析,预计内存需求80G(12万个细胞):
gsmap quick_mode \
--workdir './example_quick_mode/Mouse_Embryo' \ # 输出目录
--homolog_file 'gsMap_resource/homologs/mouse_human_homologs.txt' \ # 同源基因文件
--sample_name 'E16.5_E1S1.MOSTA' \ # 样本名称
--gsMap_resource_dir 'gsMap_resource' \ # 资源目录
--hdf5_path 'gsMap_example_data/ST/E16.5_E1S1.MOSTA.h5ad' \ # ST数据路径
--annotation 'annotation' \ # 细胞类型注释列名
--data_layer 'count' \ # 基因表达矩阵层
--sumstats_file 'gsMap_example_data/GWAS/IQ_NG_2018.sumstats.gz' \ # GWAS文件
--trait_name 'IQ' # 性状名称
输出结果路径结构如下:
example_quick_mode/Mouse_Embryo
├── E16.5_E1S1.MOSTA
│ ├── find_latent_representations # 潜在空间表征(h5ad格式)
│ ├── latent_to_gene # 基因标记评分
│ ├── generate_ldscore # LD评分矩阵
│ ├── spatial_ldsc # 空间细胞-性状关联结果
│ ├── cauchy_combination # 区域/细胞类型级关联结果
│ └── report # 交互式网页报告
关键输出文件:
-
空间关联结果:spatial_ldsc目录中的统计显著性文件(如P值矩阵)。
-
可视化报告:report目录下的HTML文件,包含空间分布热图、基因得分等。
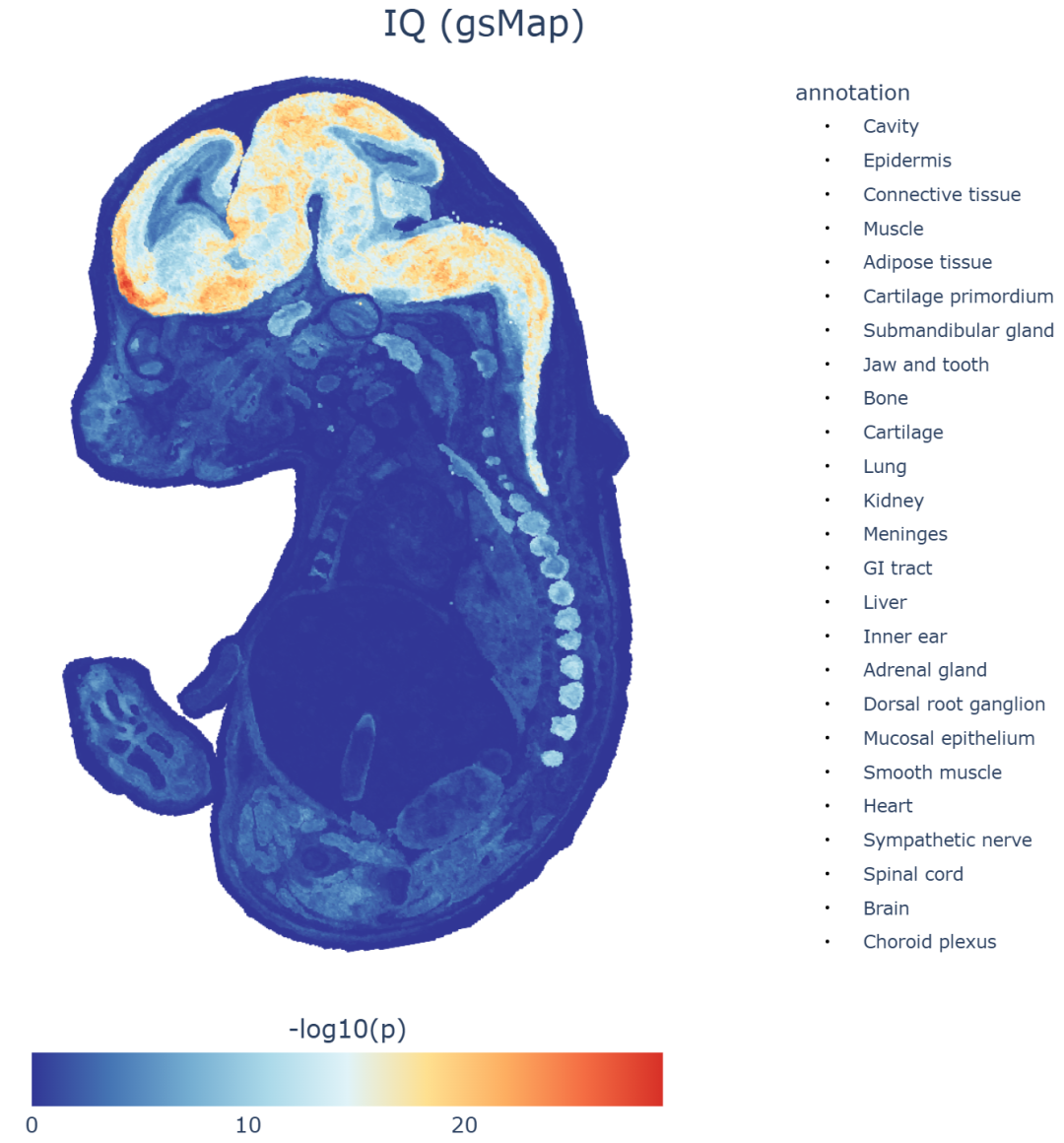
▲ IQ性状的示例分析热图结果展示。
除了上述单个性状的分析,如果你有多个性状的多个GWAS汇总数据,可以通过配置yaml文件执行批量分析:
#gwas_config.yaml文件内容如下:
Height: gsMap_example_data/GWAS/GIANT_EUR_Height_2022_Nature.sumstats.gz
IQ: gsMap_example_data/GWAS/IQ_NG_2018.sumstats.gz
SCZ: gsMap_example_data/GWAS/PGC3_SCZ_wave3_public_INFO80.sumstats.gz
运行的话只需要在上面代码的基础上使用--sumstats_config_file替换掉--sumstats_file参数:
gsmap quick_mode \
--sumstats_config_file 'gsMap_example_data/GWAS/gwas_config.yaml' # 替换--sumstats_file
多组学叠加工作量的神器
立马收藏学起来
欢迎各位佬哥佬姐关注

















 831
831

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









